Genetics of Prostate Cancer (PDQ®): Genetics - Health Professional Information [NCI]
This information is produced and provided by the National Cancer Institute (NCI). The information in this topic may have changed since it was written. For the most current information, contact the National Cancer Institute via the Internet web site at http://cancer.gov or call 1-800-4-CANCER.
This executive summary reviews the topics covered in Genetics of Prostate Cancer and provides hyperlinks to detailed sections that describe available evidence on each topic.
- Introduction
Prostate cancer is highly heritable. Up to 60% of prostate cancer risk is caused by inherited factors. This inherited risk is comprised of risk from common genetic variants and risk from pathogenic variants in moderate-risk and high-risk genes.
- Risk Factors for Prostate Cancer
Risk factors for prostate cancer include age, a family history of prostate cancer and other cancers, genetics, and ancestry (such as West African ancestry).
- Risk Assessment for Prostate Cancer
Risk assessment for prostate cancer primarily includes intake of an individual's personal cancer history, family cancer history, and ancestry. These factors are then incorporated into recommendations for prostate cancer screening.
- Indications for Prostate Cancer Germline Genetic Testing
Hereditary prostate cancer genetic testing criteria are based on one or more of the following: an individual's family history and/or genetic test results, personal/disease characteristics, and tumor sequencing results. Criteria for prostate cancer genetic testing vary based on current guidelines and expert opinion.
- Genetic Testing Approach for Prostate Cancer
Since next-generation sequencing (NGS) has become readily available and patent restrictions have been eliminated, several clinical laboratories offer multigene panel testing at a cost that is comparable to that of single-gene testing.
- Germline Genetics for Prostate Cancer
The bulk of inherited prostate cancer risk is conferred by hundreds of genetic polymorphisms, which are common in the general population. Each of these polymorphisms provides a slight increase in prostate cancer risk. For a subset of individuals, prostate cancer risk is caused by rare, deleterious variants located in specific genes.
- Prostate Cancer Genetics: Screening, Surveillance, and Treatment
This section focuses on the impacts of genetics on prostate cancer screening, surveillance, and treatment. Genetic test results are increasingly driving targeted therapy options and strategies for treatment in oncology.
Prostate cancer is highly heritable. Up to 60% of prostate cancer risk is caused by inherited factors.[1,2] The inherited risk is comprised of risk from common genetic variants and risk from pathogenic variants in moderate-risk and high-risk genes. As with breast and colon cancers, familial clustering of prostate cancer has been reported frequently.[3]
Prostate cancer clusters with particular intensity in some families. Highly to moderately penetrant genetic variants are thought to be associated with prostate cancer risk in these families. Members of these families may benefit from genetic counseling. Additionally, polygenic risk scores derived from combinations of single nucleotide polymorphisms, in addition to other risk factors like family history, race, and age/stage of prostate cancer diagnosis, have also been developed.[4,5] Recommendations and guidelines for genetic counseling referrals are based on an individual's age at prostate cancer diagnosis, prostate cancer stage at diagnosis, and specific patterns of cancer in the family history.[6,7] However, uptake of genetic testing based on an individual's family history of prostate cancer and/or a diagnosis of prostate cancer is variably implemented across practice settings and geographical regions.[8,9,10] For more information about genetic testing criteria for prostate cancer, see Table 2.
References:
- Houlahan KE, Livingstone J, Fox NS, et al.: A polygenic two-hit hypothesis for prostate cancer. J Natl Cancer Inst 115 (4): 468-472, 2023.
- Mucci LA, Hjelmborg JB, Harris JR, et al.: Familial Risk and Heritability of Cancer Among Twins in Nordic Countries. JAMA 315 (1): 68-76, 2016.
- Seibert TM, Garraway IP, Plym A, et al.: Genetic Risk Prediction for Prostate Cancer: Implications for Early Detection and Prevention. Eur Urol 83 (3): 241-248, 2023.
- Pagadala MS, Lynch J, Karunamuni R, et al.: Polygenic risk of any, metastatic, and fatal prostate cancer in the Million Veteran Program. J Natl Cancer Inst 115 (2): 190-199, 2023.
- Huynh-Le MP, Karunamuni R, Fan CC, et al.: Prostate cancer risk stratification improvement across multiple ancestries with new polygenic hazard score. Prostate Cancer Prostatic Dis 25 (4): 755-761, 2022.
- National Comprehensive Cancer Network: NCCN Clinical Practice Guidelines in Oncology: Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic. Version 2.2024. Plymouth Meeting, Pa: National Comprehensive Cancer Network, 2023. Available online with free registration. Last accessed October 31, 2023.
- National Comprehensive Cancer Network: NCCN Clinical Practice Guidelines in Oncology: Prostate Cancer Early Detection. Version 2.2023. Plymouth Meeting, Pa: National Comprehensive Cancer Network, 2023. Available online with free registration. Last accessed November 30, 2023.
- Clark NM, Flanagan MR: ASO Author Reflections: Low Genetic Testing Utilization Among Patients with Breast, Ovarian, Pancreatic, and Prostate Cancers. Ann Surg Oncol 30 (3): 1327-1328, 2023.
- Giri VN, Morgan TM, Morris DS, et al.: Genetic testing in prostate cancer management: Considerations informing primary care. CA Cancer J Clin 72 (4): 360-371, 2022.
- Russo J, Giri VN: Germline testing and genetic counselling in prostate cancer. Nat Rev Urol 19 (6): 331-343, 2022.
Age
Prostate cancer risk correlates with age. Prostate cancer is rarely seen in men younger than 40 years. The incidence rises rapidly with each decade thereafter. For example, the probability of being diagnosed with prostate cancer is 1 in 449 for men aged 49 years or younger, 1 in 26 for men aged 50 through 64 years, 1 in 10 for men aged 65 through 84 years, and 1 in 32 for men aged 85 years and older. Lifetime risk of developing prostate cancer is 1 in 8.[1] Approximately 10% of prostate cancer cases are diagnosed in men younger than 56 years and represent early-onset prostate cancer. Data from the Surveillance, Epidemiology, and End Results (SEER) Program show that early-onset prostate cancer diagnosis rates are increasing, and there is evidence that cases may be more aggressive in this subpopulation.[2]
Ancestry
The risk of developing prostate cancer is dramatically higher in Black American individuals, who predominantly have West African ancestry (186.1 cases/100,000 men) when compared with other racial and ethnic groups in the United States:
- White: 110.7 cases/100,000 men.
- Asian American or Pacific Islander: 60.9 cases/100,000 men.
- American Indian or Alaska Native: 91.9 cases/100,000 men.
- Hispanic or Latino: 90.9 cases/100,000 men.[1]
Prostate cancer mortality rates in Black individuals (37.9/100,000 men) are higher than those in other racial and ethnic groups in the United States:
- White: 17.9/100,000 men.
- Asian American or Pacific Islander: 8.7/100,000 men.
- American Indian or Alaska Native: 22.5/100,000 men.
- Hispanic or Latino: 15.4/100,000 men.[1]
Globally, prostate cancer incidence and mortality rates also vary widely from country to country.[3] The etiology of this variation in prostate cancer risk is likely multifactorial and may be due to biological factors, access to health care, and other social determinants of health.[4,5]
Family History of Prostate Cancer and Other Cancers
Results from several large case-control studies and cohort studies representing various populations suggest that family history is a major risk factor in prostate cancer.[6,7,8,9,10] A family history of a brother or father with prostate cancer increases the risk of prostate cancer, and the risk is inversely related to the age of the affected relative.[7,8,11,12,13] Risk is increased when a first-degree relative (FDR) was diagnosed with prostate cancer before age 65 years.
A meta-analysis of 33 epidemiological case-control and cohort-based studies has provided detailed information regarding risk ratios related to family history of prostate cancer (for more information, see Table 1).[14]
| Risk Group | RR for Prostate Cancer (95% CI) |
|---|---|
| CI = confidence interval; FDR = first-degree relative. | |
| a Adapted from Kiciński et al.[14] | |
| Brother(s) with prostate cancer diagnosed at any age | 3.14 (2.37–4.15) |
| Father with prostate cancer diagnosed at any age | 2.35 (2.02–2.72) |
| One affected FDR diagnosed at any age | 2.48 (2.25–2.74) |
| Affected FDRs diagnosed <65 y | 2.87 (2.21–3.74) |
| Affected FDRs diagnosed ≥65 y | 1.92 (1.49–2.47) |
| Second-degree relativesdiagnosed at any age | 2.52 (0.99–6.46) |
| Two or more affected FDRs diagnosed at any age | 4.39 (2.61–7.39) |
A family history of breast cancer is also associated with increased prostate cancer risk. In the Health Professionals Follow-up Study (HPFS), comprising over 40,000 men, those with a family history of breast cancer had a 21% higher risk of developing prostate cancer overall and a 34% increased risk of developing a lethal form of prostate cancer.[10] This is consistent with findings from previous cohorts,[15] though, notably, not all series have detected this association.[16,17] The HPFS and other studies have also shown that men with a family history of both prostate and breast/ovarian cancers were at an even higher risk of prostate cancer compared with men with a family history of either prostate or breast/ovarian cancer alone.[10,16] A proportion of the increased prostate cancer risk associated with family history of breast cancer is likely due to pathogenic variants in the DNA damage repair pathway, most commonly BRCA2.[18,19,20,21] For more information, see the BRCA1 and BRCA2 section. The association between prostate and breast cancers in families appears bidirectional. Among women, a family history of prostate cancer is likewise associated with increased risk of breast cancer.[22,23]
An association also exists between prostate cancer risk and colon cancer. Men with germline variants in DNA mismatch repair genes are at increased risk of developing prostate cancer.[24] One study reported an approximately twofold increased risk of prostate cancer among first- and second-degree relatives of probands with colorectal cancer meeting Amsterdam I or Amsterdam II criteria for Lynch syndrome.[25] For more information on Amsterdam criteria, see the Defining Lynch syndrome families section in Genetics of Colorectal Cancer.
Family history has been shown to be a risk factor for men of different races and ethnicities. In a population-based case-control study of prostate cancer among African American, White, and Asian American individuals in the United States (Los Angeles, San Francisco, and Hawaii) and Canada (Vancouver and Toronto),[26] 5% of controls and 13% of all cases reported a father, brother, or son with prostate cancer. These prevalence estimates were somewhat lower among Asian American individuals than among African American or White individuals. A positive family history was associated with a twofold to threefold increase in relative risk (RR) in each of the three ethnic groups. The overall odds ratio (OR) associated with a family history of prostate cancer was 2.5 (95% confidence interval [CI], 1.9–3.3) with adjustment for age and ethnicity.[26]
Evidence shows that a family history of prostate cancer can be associated with inferior clinical outcomes. When patients were referred for prostate biopsy (typically due to elevated prostate-specific antigen [PSA]), men with a family history of the disease were at increased risk for high-grade prostate cancer when compared with patients without a family history.[27] A large population-based study from Utah reported that men with either of the following were at an increased risk for early-onset prostate cancer: 1) three or more FDRs diagnosed with prostate cancer, or 2) two or more FDRs or second-degree relatives with prostate cancer.[28]
Genetics
There are multiple germline pathogenic variants and single nucleotide variants that are associated with prostate cancer risk. For more information about these genetic variants, see the National Human Genome Research Institute's GWAS catalog. Germline genetic testing may be indicated to assess prostate cancer risk and/or inform therapeutic decision-making in men diagnosed with prostate cancer. Prostate cancer risks vary depending on the specific gene and pathogenic variant involved.[29] Prostate cancer heritability (when considering low, moderate, and high-penetrant genetic factors) can be as high 57% (95% CI, 51%–63%).[30] Genetic variants that contribute to this risk are continuously being identified.[28] Prostate cancer heritability rates may also vary in different racial and ethnic populations.[31] For more information, see the Germline Genetics for Prostate Cancer section.
References:
- American Cancer Society: Cancer Facts and Figures 2024. American Cancer Society, 2024. Available online. Last accessed June 21, 2024.
- Salinas CA, Tsodikov A, Ishak-Howard M, et al.: Prostate cancer in young men: an important clinical entity. Nat Rev Urol 11 (6): 317-23, 2014.
- Sung H, Ferlay J, Siegel RL, et al.: Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin 71 (3): 209-249, 2021.
- Krimphove MJ, Cole AP, Fletcher SA, et al.: Evaluation of the contribution of demographics, access to health care, treatment, and tumor characteristics to racial differences in survival of advanced prostate cancer. Prostate Cancer Prostatic Dis 22 (1): 125-136, 2019.
- Fletcher SA, Marchese M, Cole AP, et al.: Geographic Distribution of Racial Differences in Prostate Cancer Mortality. JAMA Netw Open 3 (3): e201839, 2020.
- Carter BS, Beaty TH, Steinberg GD, et al.: Mendelian inheritance of familial prostate cancer. Proc Natl Acad Sci U S A 89 (8): 3367-71, 1992.
- Grönberg H, Damber L, Damber JE: Familial prostate cancer in Sweden. A nationwide register cohort study. Cancer 77 (1): 138-43, 1996.
- Cannon L, Bishop DT, Skolnick M, et al.: Genetic epidemiology of prostate cancer in the Utah Mormon genealogy. Cancer Surv 1 (1): 47-69, 1982.
- Saarimäki L, Tammela TL, Määttänen L, et al.: Family history in the Finnish Prostate Cancer Screening Trial. Int J Cancer 136 (9): 2172-7, 2015.
- Barber L, Gerke T, Markt SC, et al.: Family History of Breast or Prostate Cancer and Prostate Cancer Risk. Clin Cancer Res 24 (23): 5910-5917, 2018.
- Ghadirian P, Howe GR, Hislop TG, et al.: Family history of prostate cancer: a multi-center case-control study in Canada. Int J Cancer 70 (6): 679-81, 1997.
- Stanford JL, Ostrander EA: Familial prostate cancer. Epidemiol Rev 23 (1): 19-23, 2001.
- Matikaine MP, Pukkala E, Schleutker J, et al.: Relatives of prostate cancer patients have an increased risk of prostate and stomach cancers: a population-based, cancer registry study in Finland. Cancer Causes Control 12 (3): 223-30, 2001.
- Kiciński M, Vangronsveld J, Nawrot TS: An epidemiological reappraisal of the familial aggregation of prostate cancer: a meta-analysis. PLoS One 6 (10): e27130, 2011.
- Cerhan JR, Parker AS, Putnam SD, et al.: Family history and prostate cancer risk in a population-based cohort of Iowa men. Cancer Epidemiol Biomarkers Prev 8 (1): 53-60, 1999.
- Kalish LA, McDougal WS, McKinlay JB: Family history and the risk of prostate cancer. Urology 56 (5): 803-6, 2000.
- Damber L, Grönberg H, Damber JE: Familial prostate cancer and possible associated malignancies: nation-wide register cohort study in Sweden. Int J Cancer 78 (3): 293-7, 1998.
- Agalliu I, Karlins E, Kwon EM, et al.: Rare germline mutations in the BRCA2 gene are associated with early-onset prostate cancer. Br J Cancer 97 (6): 826-31, 2007.
- Edwards SM, Kote-Jarai Z, Meitz J, et al.: Two percent of men with early-onset prostate cancer harbor germline mutations in the BRCA2 gene. Am J Hum Genet 72 (1): 1-12, 2003.
- Ford D, Easton DF, Bishop DT, et al.: Risks of cancer in BRCA1-mutation carriers. Breast Cancer Linkage Consortium. Lancet 343 (8899): 692-5, 1994.
- Gayther SA, de Foy KA, Harrington P, et al.: The frequency of germ-line mutations in the breast cancer predisposition genes BRCA1 and BRCA2 in familial prostate cancer. The Cancer Research Campaign/British Prostate Group United Kingdom Familial Prostate Cancer Study Collaborators. Cancer Res 60 (16): 4513-8, 2000.
- Beebe-Dimmer JL, Yee C, Cote ML, et al.: Familial clustering of breast and prostate cancer and risk of postmenopausal breast cancer in the Women's Health Initiative Study. Cancer 121 (8): 1265-72, 2015.
- Sellers TA, Potter JD, Rich SS, et al.: Familial clustering of breast and prostate cancers and risk of postmenopausal breast cancer. J Natl Cancer Inst 86 (24): 1860-5, 1994.
- Dominguez-Valentin M, Sampson JR, Seppälä TT, et al.: Cancer risks by gene, age, and gender in 6350 carriers of pathogenic mismatch repair variants: findings from the Prospective Lynch Syndrome Database. Genet Med 22 (1): 15-25, 2020.
- Samadder NJ, Smith KR, Wong J, et al.: Cancer Risk in Families Fulfilling the Amsterdam Criteria for Lynch Syndrome. JAMA Oncol 3 (12): 1697-1701, 2017.
- Whittemore AS, Wu AH, Kolonel LN, et al.: Family history and prostate cancer risk in black, white, and Asian men in the United States and Canada. Am J Epidemiol 141 (8): 732-40, 1995.
- Clements MB, Vertosick EA, Guerrios-Rivera L, et al.: Defining the Impact of Family History on Detection of High-grade Prostate Cancer in a Large Multi-institutional Cohort. Eur Urol 82 (2): 163-169, 2022.
- Beebe-Dimmer JL, Kapron AL, Fraser AM, et al.: Risk of Prostate Cancer Associated With Familial and Hereditary Cancer Syndromes. J Clin Oncol 38 (16): 1807-1813, 2020.
- Seibert TM, Garraway IP, Plym A, et al.: Genetic Risk Prediction for Prostate Cancer: Implications for Early Detection and Prevention. Eur Urol 83 (3): 241-248, 2023.
- Mucci LA, Hjelmborg JB, Harris JR, et al.: Familial Risk and Heritability of Cancer Among Twins in Nordic Countries. JAMA 315 (1): 68-76, 2016.
- Bree KK, Hensley PJ, Pettaway CA: Germline Predisposition to Prostate Cancer in Diverse Populations. Urol Clin North Am 48 (3): 411-423, 2021.
Risk assessment for prostate cancer primarily involves the intake of a patient's family cancer history. Family history intake includes the following:
- Information about cancers* in male and female blood relatives on maternal and paternal sides of the family.
- Ages at cancer diagnoses.
- Ages of death from cancer.
- The number of relatives with metastatic prostate cancer.
- The number of relatives who died of prostate cancer.
- Information on relatives who are undergoing cancer screening, if known.
*Cancers include, but are not limited to, the following: prostate, breast, pancreas, colorectal, uterine, ovarian, upper gastrointestinal (GI), and skin cancers.
Ancestry is also an important component of the family history. Ashkenazi Jewish ancestry on either side of the family may prompt greater suspicion for founder pathogenic variants in BRCA1 and BRCA2, which could lead to increased cancer risk in a family. Men of African descent (Black men) also have a higher risk for prostate cancer. Within the United States, Black men (186.1 prostate cancer cases/100,000 men) have approximately a 68% higher incidence rate of prostate cancer than White men (110.7 prostate cancer cases/100,000 men).[1] Black men also have more than twice the rate of prostate cancer–specific death (37.9 deaths/100,000 men) than White men (17.9 deaths/100,000 men).[1] This increased prostate cancer risk may be due to challenges, including the following: 1) access to care, 2) limited awareness of prostate cancer screening programs, 3) limited engagement in prostate cancer screening/genetic testing, and 4) the presence of specific genetic markers that can increase prostate cancer risk.[2,3,4,5,6]
These familial risk factors are then incorporated into recommendations for prostate cancer screening. National guidelines recommend discussing prostate cancer screening with prostate-specific antigen (PSA) and digital rectal exam between the ages of 45 and 75 years for individuals at average risk for prostate cancer.
In contrast, prostate cancer screening is recommended to start at age 40 years for individuals in these high-risk groups:
Men of Black/African descent.
Men with germline pathogenic variants that increase prostate cancer risk.
Men who have family histories with features suggestive of hereditary cancer syndromes like the following:
- Hereditary breast and ovarian cancer syndrome: Family members with ovarian cancer, pancreatic cancer, metastatic/high-risk prostate cancer, male breast cancer, and/or breast cancer diagnosed at or before age 50 years.
- Lynch syndrome: Family members with colorectal or endometrial cancer diagnosed at or before age 50 years, ovarian cancer, pancreatic cancer, urothelial cancer, and/or upper GI cancer.
- Hereditary prostate cancer: Multiple generations with prostate cancer, deaths from prostate cancer, and/or family members with metastatic prostate cancer.[4,5,6]
The role of additional markers, such as polygenic risk scores, in prostate cancer risk assessment is evolving. Additional screening strategies, like multiparametric magnetic resonance imaging (mpMRI), are also being studied.
References:
- American Cancer Society: Cancer Facts and Figures 2024. American Cancer Society, 2024. Available online. Last accessed June 21, 2024.
- Liadi Y, Campbell T, Dike P, et al.: Prostate cancer metastasis and health disparities: a systematic review. Prostate Cancer Prostatic Dis 27 (2): 183-191, 2024.
- Nair SS, Chakravarty D, Dovey ZS, et al.: Why do African-American men face higher risks for lethal prostate cancer? Curr Opin Urol 32 (1): 96-101, 2022.
- National Comprehensive Cancer Network: NCCN Clinical Practice Guidelines in Oncology: Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic. Version 2.2024. Plymouth Meeting, Pa: National Comprehensive Cancer Network, 2023. Available online with free registration. Last accessed October 31, 2023.
- National Comprehensive Cancer Network: NCCN Clinical Practice Guidelines in Oncology: Prostate Cancer Early Detection. Version 2.2023. Plymouth Meeting, Pa: National Comprehensive Cancer Network, 2023. Available online with free registration. Last accessed November 30, 2023.
- National Comprehensive Cancer Network: NCCN Clinical Practice Guidelines in Oncology: Prostate Cancer. Version 4.2023. Plymouth Meeting, Pa: National Comprehensive Cancer Network, 2023. Available online with free registration. Last accessed November 30, 2023.
The criteria for consideration of genetic testing for prostate cancer varies depending on the current guidelines and expert opinion consensus, as summarized in Table 2.[1,2,3,4,5] Hereditary prostate cancer genetic testing criteria are based on an individual's family history, personal/disease characteristics, and tumor sequencing results. The genes recommended for genetic testing vary based on national guidelines and consensus conference recommendations. Precision therapy has emerged as a major driver for germline genetic testing and may be a separate reason to pursue testing beyond the criteria stated in Table 2. The National Comprehensive Cancer Network (NCCN) Prostate Cancer guidelines recommend testing for at least BRCA1, BRCA2, ATM, CHEK2, PALB2, HOXB13,MLH1, MSH2, MSH6, and PMS2 for men meeting specific testing indications.[4] A consensus conference in 2019 addressed the role of genetic testing for inherited prostate cancer.[6] Family history–based indications for genetic testing included testing for BRCA1/BRCA2, HOXB13, DNA mismatch repair (MMR) genes, and ATM. Tumor sequencing that identifies variants that may be germline in origin, like variants in BRCA1/BRCA2, DNA MMR genes, or ATM and other genes, warrants confirmatory germline testing. Somatic findings for which germline testing is considered include the following:
- Somatic variants that are associated with germline susceptibility.
- Hypermutated tumors, which are indicative of DNA MMR.
- Chromosome rearrangements in specific tumors.
- High-variant allele frequency (percent of sequence reads that have the identified variant). Variant allele frequency can be altered for reasons not associated with germline variants such as loss of heterozygosity, ploidy (copy number variants), tumor heterogeneity, and tumor sample purity.[7]
It is recommended that germline genetic testing candidates undergo genetic education and counseling before participating in testing. Genetic counseling provides information about genetic testing and possible testing outcomes (including risks, benefits, limitations, and familial, psychological, and health care–based implications that vary depending on results). Genetic education and counseling help individuals make informed decisions about whether they should undergo germline genetic testing. For more information on genetic education and genetic counseling, see Cancer Genetics Risk Assessment and Counseling.
| | Philadelphia Prostate Cancer Consensus Conference (Giri et al. 2020)a[6] | Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic (Version 2.2024)b[3] | NCCN Prostate Cancer (Version 4.2023)c[4] | European Advanced Prostate Cancer Consensus Conference (Gillessen et al. 2017[2]and Gillessen 2020[8])d |
|---|---|---|---|---|
| dMMR = mismatch repair deficient; FDR = first-degree relative; HBOC = hereditary breast and ovarian cancer; MMR = mismatch repair; MSI = microsatellite instability; NCCN = National Comprehensive Cancer Network; SDR= second-degree relative; TDR= third-degree relative. | ||||
| a Giri et al.: Specific genes to test includeBRCA1/BRCA2, DNA MMR genes,ATM, andHOXB13depending on various testing indications. | ||||
| b NCCN Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic guidelines state that prostate cancer risk management is indicated forBRCA1andBRCA2carriers, but evidence for risk management is insufficient for other genes. | ||||
| c NCCN Prostate Cancer guidelines specify that germline multigene testing includes at least the following genes:BRCA1,BRCA2,ATM,PALB2,CHEK2,MLH1,MSH2,MSH6, andPMS2. Including additional genes may be appropriate based on clinical context. | ||||
| d Gillessen et al. endorsed the use of large panel testing including homologous recombination and DNA MMR genes. | ||||
| Family History Criteria | All men with prostate cancer from families meeting established testing or syndromic criteria for HBOC, hereditary prostate cancer, or Lynch syndrome | Men affected with prostate cancer who have a family history of the following: ≥1 FDR,SDR, or TDR (on the same side of the family) with breast cancer at age ≤50 y or with any of the following: triple-negative breast cancer, ovarian cancer, pancreatic cancer, high- or very-high-risk prostate cancer, male breast cancer, or metastatic prostate cancer at any age | Men affected with prostate cancer who have the following: ≥1 FDR, SDR, or TDR (on the same side of the family) with breast cancer at age ≤50 y, colorectal or endometrial cancer at age ≤50 y, male breast cancer at any age, ovarian cancer at any age, exocrine pancreatic cancer at any age, or metastatic, regional, very-high-risk, high-risk prostate cancer at any age | Men with a positive family history of prostate cancer[2] |
| Men affected with prostate cancer who have >2 close biological relatives with a cancer associated with HBOC, hereditary prostate cancer, or Lynch syndrome | Men affected with prostate cancer who have ≥3 FDRs, SDRs, or TDRs (on the same side of the family) with breast cancer or prostate cancer (any grade) at any age | Men affected with prostate cancer who have ≥1 FDR with prostate cancer at age ≤60 y (exclude relatives with clinically localized Grade Group 1 disease) | Men with a positive family history of other cancer syndromes (HBOC and/or pancreatic cancer and/or Lynch syndrome)[2] | |
| Men with anFDRwho was diagnosed with prostate cancer at <60 y | Men with or without prostate cancer with an FDR who meets any of the criteria listed above (except when a man without prostate cancer has relatives who meet the above criteria solely for systemic therapy decision-making; these criteria may also be extended to an affected TDR if he/she is related to the patient through two male relatives) | Men affected with prostate cancer who have ≥2 FDRs, SDRs, or TDRs (on the same side of the family) with breast cancer or prostate cancer at any age (exclude relatives with clinically localized Grade Group 1 disease) | ||
| Men with relatives who died of prostate cancer | Men affected with prostate cancer who have ≥3 FDRs or SDRs (on the same side of the family) with the following Lynch syndrome-related cancers, especially if diagnosed at age <50 y: colorectal, endometrial, gastric, ovarian, exocrine pancreas, upper tract urothelial, glioblastoma, biliary tract, and small intestine | |||
| Men with a metastatic prostate cancer in an FDR | ||||
| Consider genetic testing in men with prostate cancer andAshkenazi Jewishancestry | Men with prostate cancer and Ashkenazi Jewish ancestry | Men with prostate cancer and Ashkenazi Jewish ancestry | ||
| Men with prostate cancer and a known family history of a pathogenic or likely pathogenic variant in one of the following genes:BRCA1,BRCA2,ATM,PALB2,CHEK2,MLH1,MSH2,MSH6,PMS2, orEPCAM | ||||
| Clinical/Pathological Features | Men with metastatic prostate cancer | Men with metastatic prostate cancer | Men with metastatic prostate cancer | Men with newly diagnosed metastatic prostate cancer (62% of panel voted in favor ofgenetic counseling /testing in a minority of selected patients)[8] |
| Men with stage T3a or higher prostate cancer | Men with high- or very-high-risk prostate cancer | Men with high-risk prostate cancer, very-high-risk prostate cancer, high-risk localized prostate cancer, or regional (node-positive) prostate cancer | ||
| Men with prostate cancer that has intraductal/ductal histology | Testing may be considered in men who have intermediate-risk prostate cancer with intraductal/cribriform histology at any age | Germline testing may be considered in men who have intermediate-risk prostate cancer with intraductal/cribriform histology at any age | ||
| Germline testing may be considered in men with prostate cancer AND a prior personal history of any of the following cancers: exocrine pancreatic, colorectal, gastric, melanoma, upper tract urothelial, glioblastoma, biliary tract, and small intestinal | Men with prostate cancer diagnosed at age <60 y[2] | |||
| Tumor Sequencing Characteristics | Men with prostate cancer whose somatic testing reveals the possibility of a germline variant in a cancer risk gene, especiallyBRCA2,BRCA1,ATM, and DNA MMR genes | Men with a pathogenic variant found on tumor genomic testing that may have clinical implications if it is also identified in the germline | Recommend tumor testing forpathogenic variantsin homologous recombination genes in men with metastatic disease; consider tumor testing in men with regional prostate cancer | |
| RecommendMSI -high or dMMR tumor testing in men with metastatic castration-resistant prostate cancer; consider testing in men with regional or castration-sensitive metastatic prostate cancer | ||||
References:
- Giri VN, Knudsen KE, Kelly WK, et al.: Role of Genetic Testing for Inherited Prostate Cancer Risk: Philadelphia Prostate Cancer Consensus Conference 2017. J Clin Oncol 36 (4): 414-424, 2018.
- Gillessen S, Attard G, Beer TM, et al.: Management of Patients with Advanced Prostate Cancer: The Report of the Advanced Prostate Cancer Consensus Conference APCCC 2017. Eur Urol 73 (2): 178-211, 2018.
- National Comprehensive Cancer Network: NCCN Clinical Practice Guidelines in Oncology: Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic. Version 2.2024. Plymouth Meeting, Pa: National Comprehensive Cancer Network, 2023. Available online with free registration. Last accessed October 31, 2023.
- National Comprehensive Cancer Network: NCCN Clinical Practice Guidelines in Oncology: Prostate Cancer. Version 4.2023. Plymouth Meeting, Pa: National Comprehensive Cancer Network, 2023. Available online with free registration. Last accessed November 30, 2023.
- National Comprehensive Cancer Network: NCCN Clinical Practice Guidelines in Oncology: Prostate Cancer Early Detection. Version 2.2023. Plymouth Meeting, Pa: National Comprehensive Cancer Network, 2023. Available online with free registration. Last accessed November 30, 2023.
- Giri VN, Knudsen KE, Kelly WK, et al.: Implementation of Germline Testing for Prostate Cancer: Philadelphia Prostate Cancer Consensus Conference 2019. J Clin Oncol 38 (24): 2798-2811, 2020.
- Raymond VM, Gray SW, Roychowdhury S, et al.: Germline Findings in Tumor-Only Sequencing: Points to Consider for Clinicians and Laboratories. J Natl Cancer Inst 108 (4): , 2016.
- Gillessen S, Attard G, Beer TM, et al.: Management of Patients with Advanced Prostate Cancer: Report of the Advanced Prostate Cancer Consensus Conference 2019. Eur Urol 77 (4): 508-547, 2020.
Since next-generation sequencing (NGS) has become readily available and patent restrictions have been eliminated, several clinical laboratories offer multigene panel testing at a cost that is comparable to that of single-gene testing. Three types of genetic test results can be reported: 1) pathogenic/likely pathogenic variants, 2) variants of uncertain significance (VUS), or 3) negative results. Patients need pretest genetic counseling or informed consent to understand germline genetic testing results. For example, patients should understand that VUS can be reported, that VUS do not immediately impact care/inform cancer risk, and that VUS may be reclassified as either pathogenic/likely pathogenic or benign/likely benign when more data are acquired. For more information on genetic counseling considerations and research associated with multigene testing, see the Multigene (panel) testing section in Cancer Genetics Risk Assessment and Counseling.
Prostate cancer is highly heritable. More than half of an individual's prostate cancer risk is inherited from one's parents.[1] Considerable work has been performed to identify and characterize inherited germline variants that contribute to the genetic portion of prostate cancer risk. For most patients, the bulk of inherited risk is conferred by hundreds of genetic polymorphisms, which are common in the general population. Each of these polymorphisms slightly increases prostate cancer risk. For a small subset of patients, prostate cancer risk is generated by rare, deleterious variants located in specific genes. In this section, we will describe the specific genes implicated in inherited prostate cancer risk and the many common polymorphisms (which are typically located in the genomic space between genes) that create a risk profile for most patients.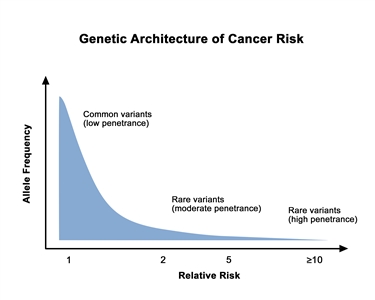
Genetic architecture of cancer risk. This graph depicts the general finding of a low relative risk associated with common, low-penetrance genetic variants, such as single-nucleotide polymorphisms identified in genome-wide association studies, and a higher relative risk associated with rare, high-penetrance genetic variants, such as mutations in the BRCA1/ BRCA2 genes associated with hereditary breast and ovarian cancer and the mismatch repair genes associated with Lynch syndrome.
Clinically Relevant Genes for Prostate Cancer
BRCA1andBRCA2
Studies of male carriers of BRCA1 and BRCA2 pathogenic variants demonstrate that these individuals have a higher risk of prostate cancer and other cancers.[2,3] Prostate cancer, in particular, has been observed at higher rates in male carriers of BRCA2 pathogenic variants than in the general population.[4] For more information about BRCA1 and BRCA2 pathogenic variants, see BRCA1 and BRCA2: Cancer Risks and Management.
BRCA–associated prostate cancer risk
The risk of prostate cancer in carriers of BRCA pathogenic variants has been studied in various settings.
In an effort to clarify the relationship between BRCA pathogenic variants and prostate cancer risk, findings from a systematic review and meta-analysis are summarized in Table 3 .
| Population | Number of Studies | Fixed-Effect Pooled Prostate Cancer RR (95% CI) | Random-Effect Pooled Prostate Cancer RR (95% CI) | I2 |
|---|---|---|---|---|
| CI = confidence interval; RR = relative risk. | ||||
| a Adapted from Nyberg et al. | ||||
| BRCA1 | ||||
| All | 20 | 1.57 (1.30–1.91) | 1.69 (1.30–2.20) | 30% |
| Unselected for age, aggressive prostate cancer, or prostate cancer family history | 15 | 1.43 (1.71–1.75) | 1.47 (1.13–1.91) | 25% |
| Unselected for age, aggressive prostate cancer, or prostate cancer family historyand did not use historical controls | 13 | 1.32 (1.07–1.64) | 1.33 (1.05–1.69) | 8% |
| Prostate cancer diagnosed <65 y | 4 | 2.21 (1.47–3.30) | 2.19 (1.21–3.98) | 57% |
| Prostate cancer diagnosed >65 y | 3 | 1.18 (0.83–1.70) | 1.43 (0.71–2.87) | 65% |
| BRCA2 | ||||
| All | 21 | 5.24 (4.63–5.49) | 3.94 (2.79–5.56) | 83% |
| Unselected for age, aggressive prostate cancer, or prostate cancer family history | 15 | 3.87 (3.34–4.47) | 3.33 (2.57–4.33) | 58% |
| Prostate cancer diagnosed <65 y | 5 | 6.37 (4.81–8.43) | 5.28 (3.10–9.00) | 63% |
| Prostate cancer diagnosed >65 y | 3 | 3.74 (2.82–4.96) | 3.74 (2.82–4.96) | 0% |
Prevalence ofBRCAfounder pathogenic variants in men with prostate cancer
Ashkenazi Jewish population
Several studies in Israel and in North America have analyzed the frequency of BRCAfounder pathogenic variants among Ashkenazi Jewish (AJ) men with prostate cancer.[5,6,7] Two specific BRCA1 pathogenic variants (185delAG and 5382insC) and one BRCA2 pathogenic variant (6174delT) are common in individuals of AJ ancestry. Carrier frequencies for these pathogenic variants in the general Jewish population are 0.9% (95% CI, 0.7%–1.1%) for the 185delAG pathogenic variant, 0.3% (95% CI, 0.2%–0.4%) for the 5382insC pathogenic variant, and 1.3% (95% CI, 1.0%–1.5%) for the BRCA2 6174delT pathogenic variant.[8,9,10,11] In these studies, the relative risks (RRs) were commonly greater than 1, but only a few were statistically significant. Many of these studies were not sufficiently powered to rule out a lower, but clinically significant, risk of prostate cancer in carriers of Ashkenazi BRCA founder pathogenic variants.
Table 4 summarizes the findings from a systemic review and meta-analysis, which help clarify the relationship between BRCA pathogenic variants and prostate cancer risk in individuals of Ashkenazi Jewish heritage.
| Population | Number of Studies | Fixed-Effect Pooled Prostate Cancer RR (95% CI) | Random-Effect Pooled Prostate Cancer RR (95% CI) | I2 |
|---|---|---|---|---|
| CI = confidence interval; RR = relative risk. | ||||
| a Adapted from Nyberg et al. | ||||
| BRCA1 | ||||
| All | 3 | 1.12 (0.55–2.31) | 1.12 (0.55–2.31) | 0% |
| BRCA2 | ||||
| All | 6 | 2.08 (1.38–3.12) | 2.08 (1.38–3.12) | 0% |
This systematic review and meta-analysis provide further evidence that prostate cancer occurs more often in Ashkenazi Jewish BRCA founder variant carriers and suggests that prostate cancer risk may be greater in men with BRCA2 6174delT founder pathogenic variants than in men with BRCA1 85delAG or BRCA1 5382insC founder pathogenic variants.
Other populations
The association between prostate cancer and pathogenic variants in BRCA1 and BRCA2 has also been studied in other populations. Table 5 summarizes studies from a systematic review and meta-analysis. This table reports the prevalence of BRCA pathogenic variants in men with prostate cancer from other varied populations.
| Population | Number of Studies | Fixed-Effect Pooled Prostate Cancer RR (95% CI) | Random-Effect Pooled Prostate Cancer RR (95% CI) | I2 |
|---|---|---|---|---|
| CI = confidence interval; RR = relative risk. | ||||
| a Adapted from Nyberg et al. | ||||
| BRCA1 | ||||
| Non-Ashkenazi European Ancestry | 8 | 1.30 (1.03–1.64) | 1.30 (0.95–1.79) | 30% |
| African Ancestry | 1 | 1.11 (0.09–13.61) | 1.11 (0.09–13.61) | - |
| Asian Ancestry | 1 | 2.27 (0.92–5.59) | 2.27 (0.92–5.59) | - |
| BRCA2 | ||||
| Non-Ashkenazi European Ancestry | 7 | 4.07 (3.45–4.80) | 3.69 (2.71–5.04) | 66% |
| African Ancestry | 1 | 10.30 (1.28–82.73) | 10.30 (1.28–82.73) | - |
| Asian Ancestry | 1 | 5.65 (3.49–9.15) | 5.65 (3.49–9.15) | - |
Prostate cancer aggressiveness in carriers ofBRCApathogenic variants
A systematic review and meta-analysis found that BRCA1 and BRCA2 showed differences in prostate cancer aggressiveness.[3] The pooled, random-effects RRs of aggressive prostate cancer (using any definition of aggressiveness) were the following for BRCA1 and BRCA2:
- BRCA1: RR, 1.98 (1.35–2.90; I² = 0%).
- BRCA2: RR, 6.08 (3.44–10.8; I² = 82%).
Men harboring pathogenic variants in the United Kingdom and Ireland were prospectively followed for prostate cancer diagnoses (BRCA1 [n = 16/376] and BRCA2 [n = 26/447]; median follow-up, 5.9 y and 5.3 y, respectively).[12] The prostate cancers identified covered the spectrum of Gleason scores from less than 6 to greater than 8; however, they differed by gene:
- BRCA1 Gleason score less than 6; standardized incidence ratio (SIR), 3.50 (95% CI, 1.67–7.35) and Gleason score greater than 7; SIR, 1.80 (95% CI, 0.89–3.65).
- BRCA2 Gleason score less than 6; SIR, 3.03 (95% CI, 1.24–7.44) and Gleason score greater than 7; SIR, 5.07 (95% CI, 3.20–8.02).
This study was followed by a large, retrospective, international study of men diagnosed with prostate cancer who had pathogenic variants in BRCA1 (n = 3,453) and BRCA2 (n = 3,051).[13] In BRCA1, there were no statistically significant associations between overall prostate cancer risk/prostate cancer with a Gleason score of 8 or higher and pathogenic sequence variant types, pathogenic variant function, or the region of the gene in which a pathogenic variant occurred, such as RING or BRCA1 C-terminal (BRCT) domains. In contrast, two prostate cancer cluster regions were identified in BRCA2: 1) 3' of BRCA2 c.7914 (hazard ratio [HR],1.78; 95% confidence interval [CI], 1.25–2.52; P = .001), and 2) BRCA2 c.756–c.1000 (HR, 2.83; 95% CI, 1.71–4.68; P = 4.0 x 10-5).
These studies suggest that prostate cancer in BRCA carriers is associated with aggressive disease features including a high Gleason score, and a high tumor stage and/or grade at diagnosis. This is a finding that warrants consideration when patients undergo cancer risk assessment and genetic counseling.[14] Research is under way to gain insight into the biological basis of aggressive prostate cancer in carriers of BRCA pathogenic variants. One study of 14 BRCA2 germline pathogenic variant carriers reported that BRCA2-associated prostate cancers harbor increased genomic instability and a mutational profile that more closely resembles metastatic prostate cancer than localized disease, with genomic and epigenomic dysregulation of the MED12L/MED12 axis similar to metastatic castration-resistant prostate cancer.[15]
BRCA1/BRCA2and survival outcomes
Analyses of prostate cancer cases in families with known BRCA1 or BRCA2 pathogenic variants have been examined for survival. A meta-analysis that examined BRCA1/BRCA2 prostate cancer risk, BRCA1/BRCA2 frequency in patients with prostate cancer, and prostate cancer mortality found that BRCA1/BRCA2 carriers who were diagnosed with prostate cancer had decreased cancer-specific survival (HR, 2.53; 95% CI, 1.98–3.22; P < .0001) when compared with noncarriers.[16] Similarly, prostate cancer overall survival (OS) was lower in men with BRCA1/BRCA2 pathogenic variants (HR, 2.08; 95% CI, 1.55–2.79; P < .0001). BRCA2 carriers had decreased cancer-specific survival (HR, 2.63; 95% CI, 2.00–3.47; P < .0001) and OS (HR, 2.21; 95% CI, 1.64–2.99; P < .0001) values when compared with noncarriers. BRCA2 carriers (BRCA2, 71.1%; 95% CI, 31.4%–93.0%) were also more likely to have prostate cancer with a Gleason score of 7 or greater than BRCA1 carriers (BRCA1, 36.3%; 95% CI, 20.0%–56.5%).
HOXB13
Key points
HOXB13 was the first gene found to be associated with hereditary prostate cancer. The HOXB13 G84E variant has been extensively studied because of its association with prostate cancer risk.
- Overall risk of prostate cancer with the G84E variant ranges from 3- to 5-fold, with a higher risk of early-onset prostate cancer with the G84E variant of up to 10-fold.
- Penetrance for carriers of the G84E variant is an approximate 60% lifetime risk of prostate cancer by age 80 years.
- There is no clear association of the G84E variant with aggressive prostate cancer or other cancers.
- Preliminary studies suggest additional variants in HOXB13 may be relevant for prostate cancer risk in diverse populations.
Background
Linkage to 17q21-22 was initially reported by the UM-PCGP from 175 pedigrees of families with hereditary prostate cancer.[17] Fine-mapping of this region provided strong evidence of linkage (LOD score, 5.49) and a narrow candidate interval (15.5 Mb) for a putative susceptibility gene among 147 families with four or more affected men and average age at diagnosis of 65 years or younger.[18] The exons of 200 genes in the 17q21-22 region were sequenced in DNA from 94 unrelated patients from hereditary prostate cancer families (from the UM-PCGP and Johns Hopkins University).[19]Probands from four families were discovered to have a recurrent pathogenic variant (G84E) in HOXB13, and 18 men with prostate cancer from these four families carried the pathogenic variant. The pathogenic variant status was determined in 5,083 additional cases and 2,662 controls. Carrier frequencies and ORs for prostate cancer risk were as follows:
- Men with a positive family history of prostate cancer, 2.2% versus negative, 0.8% (OR, 2.8; 95% CI, 1.6–5.1; P = 1.2 × 10-4).
- Men younger than 55 years at diagnosis, 2.2% versus older than 55 years, 0.8% (OR, 2.7; 95% CI, 1.6–4.7; P = 1.1 × 10-4).
- Men with a positive family history of prostate cancer and younger than 55 years at diagnosis, 3.1% versus a negative family history of prostate cancer and age at diagnosis older than 55 years, 0.6% (OR, 5.1; 95% CI, 2.4–12.2; P = 2.0 × 10-6).
- Men with a positive family history of prostate cancer and older than 55 years at diagnosis, 1.2%.
- Controls, 0.1% to 0.2%.[19]
The clinical utility of genetic testing for the HOXB13 G84E variant is evolving.[20,21]
Validation and confirmatory studies
A validation study from the International Consortium of Prostate Cancer Genetics confirmed HOXB13 as a susceptibility gene for prostate cancer risk.[22] Within carrier families, the G84E pathogenic variant was more common among men with prostate cancer than among unaffected men (OR, 4.42; 95% CI, 2.56–7.64). The G84E pathogenic variant was also significantly overtransmitted from parents to affected offspring (P = 6.5 × 10-6).
Additional studies have emerged that better define the carrier frequency and prostate cancer risk associated with the HOXB13 G84E pathogenic variant.[19,23,24,25,26,27,28] This pathogenic variant appears to be restricted to White men, primarily of European descent.[19,23,24,25] The highest carrier frequency of 6.25% was reported in Finnish early-onset cases.[26] A pooled analysis of European Americans that included 9,016 cases and 9,678 controls found an overall G84E pathogenic variant frequency of 1.34% among cases and 0.28% among controls.[27]
Risk of prostate cancer by HOXB13 G84E pathogenic variant status has been reported to vary by age of onset, family history, and geographical region. A validation study in an independent cohort of 9,988 cases and 61,994 controls from six studies of men of European ancestry, including 4,537 cases and 54,444 controls from Iceland whose genotypes were largely imputed, reported an OR of 7.06 (95% CI, 4.62–10.78; P = 1.5 × 10−19) for prostate cancer risk by G84E carrier status.[29] A pooled analysis reported a prostate cancer OR of 4.86 (95% CI, 3.18–7.69; P = 3.48 × 10-17) in men with HOXB13 pathogenic variants compared with noncarriers; this increased to an OR of 8.41 (95% CI, 5.27–13.76; P = 2.72 ×10-22) among men diagnosed with prostate cancer at age 55 years or younger. The OR was 7.19 (95% CI, 4.55–11.67; P = 9.3 × 10-21) among men with a positive family history of prostate cancer and 3.09 (95% CI, 1.83–5.23; P = 6.26 × 10-6) among men with a negative family history of prostate cancer.[27] A meta-analysis that included 24,213 cases and 73,631 controls of European descent revealed an overall OR for prostate cancer by carrier status of 4.07 (95% CI, 3.05–5.45; P < .00001). Risk of prostate cancer varied by geographical region: United States (OR, 5.10; 95% CI, 3.21–8.10; P < .00001), Canada (OR, 5.80; 95% CI, 1.27–26.51; P = .02), Northern Europe (OR, 3.61; 95% CI, 2.81–4.64; P < .00001), and Western Europe (OR, 8.47; 95% CI, 3.68–19.48; P < .00001).[24] In addition, the association between the G84E pathogenic variant and prostate cancer risk was higher for early-onset cases (OR, 10.11; 95% CI, 5.97–17.12). There was no significant association with aggressive disease in the meta-analysis.
Another meta-analysis that included 11 case-control studies also reported higher risk estimates for prostate cancer in HOXB13 G84E carriers (OR, 4.51; 95% CI, 3.28–6.20; P < .00001) and found a stronger association between HOXB13 G84E and early-onset disease (OR, 9.73; 95% CI, 6.57–14.39; P < .00001).[30] An additional meta-analysis of 25 studies that included 51,390 cases and 93,867 controls revealed an OR for prostate cancer of 3.248 (95% CI, 2.121–3.888). The association was most significant in White individuals (OR, 2.673; 95% CI, 1.920–3.720), especially those of European descent. No association was found for breast or colorectal cancer.[31] One population-based, case-control study from the United States confirmed the association of the G84E pathogenic variant with prostate cancer (OR, 3.30; 95% CI, 1.21–8.96) and reported a suggestive association with aggressive disease.[32] In addition, one study identified no men of AJ ancestry who carried the G84E pathogenic variant.[33] A case-control study from the United Kingdom that included 8,652 cases and 5,252 controls also confirmed the association of HOXB13 G84E with prostate cancer (OR, 2.93; 95% CI, 1.94–4.59; P = 6.27 × 10-8).[34] The risk was higher among men with a family history of the disease (OR, 4.53; 95% CI, 2.86–7.34; P = 3.1 × 10−8) and in early-onset prostate cancer (diagnosed at age 55 y or younger) (OR, 3.11; 95% CI, 1.98–5.00; P = 6.1 × 10−7). No association was found between carrier status and Gleason score, cancer stage, OS, or cancer-specific survival.
However, a 2018 publication of a study combining multiple prostate cancer cases and controls of Nordic origin along with functional analysis reported that simultaneous presence of HOXB13 (G84E) and CIP2A (R229Q) predisposes men to an increased risk of prostate cancer (OR, 21.1; P = .000024).[35] Furthermore, dual carriers had elevated risk for high Gleason score (OR, 2.3; P = .025) and worse prostate cancer–specific survival (hazard ratio [HR], 3.9; P = .048). Clinical validation is needed.
HOXB13pathogenic variants in diverse populations
A study of Chinese men with and without prostate cancer failed to identify the HOXB13 G84E pathogenic variant; however, there was an excess of a novel variant, G135E, in cases compared with controls.[36] A large study of approximately 20,000 Japanese men with and without prostate cancer identified another novel HOXB13 variant, G132E, which was associated with prostate cancer with an OR of 6.08 (95% CI, 3.39–11.59).[37]
Two studies confirmed the association between the HOXB13 X285K variant and increased prostate cancer risk in African American men after this variant was identified in Martinique.[38] One of these was a single-institution study, which sequenced HOXB13 in a clinical patient population of 1,048 African American men undergoing prostatectomy for prostate cancer.[39] The HOXB13 X285K variant was identified in eight patients. In a case–case analysis, X285K variant carriers were at increased risk of developing clinically significant prostate cancer (1.2% X285K carrier rate in prostate cancers with a Gleason score ≥7 vs. 0% X285K carrier rate in prostate cancers with Gleason score <7; P = .028). Similarly, X285K variant carriers also had an increased chance of developing prostate cancer at an early age (2.4% X285K carrier rate in patients <50 years vs. 0.5% X285K carrier rate in patients ≥50 years; OR, 5.25; 95% CI, 1.00–28.52; P = .03). A second study included 11,688 prostate cancer cases and 10,673 controls from multiple large consortia.[40] The HOXB13 X285K variant was only present in men of West African ancestry and was associated with a 2.4-fold increased chance of developing prostate cancer (95% CI, 1.5–3.9; P = 2 x 10-4). Individuals with the X285K variant were also more likely to have aggressive and advanced prostate cancer (Gleason score ≥8: OR, 4.7; 95% CI, 2.3–9.5; P = 2 x 10-5; stage T3/T4: OR, 4.5; 95% CI, 2.0–10.0; P = 2 x 10-4; metastatic disease: OR, 5.1; 95% CI, 1.9–13.7; P = .001). This information is important to consider when developing genetic tests for HOXB13 pathogenic variants in broader populations.
Penetrance
Penetrance estimates for prostate cancer development in carriers of the HOXB13 G84E pathogenic variant are also being reported. One study from Sweden estimated a 33% lifetime risk of prostate cancer among G84E carriers.[41] Another study from Australia reported an age-specific cumulative risk of prostate cancer of up to 60% by age 80 years.[42] A study in the United Kingdom that included HOXB13 genotype data from nearly 12,000 men with prostate cancer enrolled between 1993 and 2014 reported that the average predicted risk of prostate cancer by age 85 years is 62% (95% CI, 47%–76%) for carriers of the G84E pathogenic variant. The risk of developing prostate cancer in variant carriers increased if the men had affected family members, especially those diagnosed at an early age.[43]
Biology
HOXB13 plays a role in prostate cancer development and interacts with the androgen receptor; however, the mechanism by which it contributes to the pathogenesis of prostate cancer remains unknown. This is the first gene identified to account for a fraction of hereditary prostate cancer, particularly early-onset prostate cancer. The clinical utility and implications for genetic counseling regarding HOXB13 G84E or other pathogenic variants have yet to be defined.
DNA mismatch repair genes (Lynch syndrome)
Five genes are implicated in mismatch repair (MMR), namely MLH1, MSH2, MSH6, PMS2, and EPCAM. Germline pathogenic variants in these five genes have been associated with Lynch syndrome, which manifests by cases of nonpolyposis colorectal cancer and a constellation of other cancers in families, including endometrial, ovarian, duodenal cancers, and transitional cell cancers of the ureter and renal pelvis. For more information about other cancers that are associated with Lynch syndrome, see the Lynch syndrome section in Genetics of Colorectal Cancer. Reports have suggested that prostate cancer may be observed in men harboring an MMR gene pathogenic variant.[44,45] The first quantitative study described nine cases of prostate cancer occurring in a population-based cohort of 106 Norwegian male carriers of MMR gene pathogenic variants or obligate carriers.[46] The expected number of cases among these 106 men was 1.52 (P < .01); the men were younger at the time of diagnosis (60.4 y vs. 66.6 y; P = .006) and had more evidence of Gleason score of 8 to 10 (P < .00001) than the cases from the Norwegian Cancer Registry. Kaplan-Meier analysis revealed that the cumulative risk of prostate cancer diagnosis by age 70 years was 30% in carriers of MMR gene pathogenic variants and 8% in the general population. This finding awaits confirmation in additional populations. A population-based case-control study examined haplotype-tagging SNVs in three MMR genes (MLH1, MSH2, and PMS2). This study provided some evidence supporting the contribution of genetic variation in MLH1 and overall risk of prostate cancer.[47] To assess the contribution of prostate cancer as a feature of Lynch syndrome, one study performed microsatellite instability (MSI) testing on prostate cancer tissue blocks from families enrolled in a prostate cancer family registry who also reported a history of colon cancer. Among 35 tissue blocks from 31 distinct families, two tumors from families with MMR gene pathogenic variants were found to be MSI-high. The authors conclude that MSI is rare in hereditary prostate cancer.[48] Other studies are attempting to characterize rates of prostate cancer in Lynch syndrome families and correlate molecular features with prostate cancer risk.[49]
One study that included two familial cancer registries found an increased cumulative incidence and risk of prostate cancer among 198 independent families with MMR gene pathogenic variants and Lynch syndrome.[50] The cumulative lifetime risk of prostate cancer (to age 80 y) was 30.0% (95% CI, 16.54%–41.30%; P = .07) in carriers of MMR gene pathogenic variants, whereas it was 17.84% in the general population, according to the Surveillance, Epidemiology, and End Results (SEER) Program estimates. There was a trend of increased prostate cancer risk in carriers of pathogenic variants by age 50 years, where the risk was 0.64% (95% CI, 0.24%–1.01%; P = .06), compared with a risk of 0.26% in the general population. Overall, the HR (to age 80 y) for prostate cancer in carriers of MMR gene pathogenic variants in the combined data set was 1.99 (95% CI, 1.31–3.03; P = .0013). Among men aged 20 to 59 years, the HR was 2.48 (95% CI, 1.34–4.59; P = .0038).
A systematic review and meta-analysis that included 23 studies (6 studies with molecular characterization and 18 risk studies, of which 12 studies quantified risk for prostate cancer) reported an association of prostate cancer with Lynch syndrome.[51] In the six molecular studies included in the analysis, 73% (95% CI, 57%–85%) of prostate cancers in carriers of MMR gene pathogenic variants were MMR deficient. The RR of prostate cancer in carriers of MMR gene pathogenic variants was estimated to be 3.67 (95% CI, 2.32–6.67). Of the twelve risk studies, the RR of prostate cancer ranged from 2.11 to 2.28, compared with that seen in the general population depending on carrier status, prior diagnosis of colorectal cancer, or unknown male carrier status from families with a known pathogenic variant.
A study from three sites participating in the Colon Cancer Family Registry examined 32 cases of prostate cancer (mean age at diagnosis, 62 y; standard deviation, 8 y) in men with a documented MMR gene pathogenic variant (23 MSH2 carriers, 5 MLH1 carriers, and 4 MSH6 carriers).[52] Seventy-two percent (n = 23) had a previous diagnosis of colorectal cancer. Immunohistochemistry was used to assess MMR protein loss, which was observed in 22 tumors (69%); the pattern of loss of protein expression was 100% concordant with the germline pathogenic variant. The RR of prostate cancer was highest in carriers of MSH2 pathogenic variants (RR, 5.8; 95% CI, 2.6–20.9); the RRs in carriers of MLH1 and MSH6 pathogenic variants were 1.7 (95% CI, 1.1–6.7) and 1.3 (95% CI, 1.1–5.3), respectively. Gleason scores ranged from 5 to 10; two tumors had a Gleason score of 5; 22 tumors had a Gleason score of 6 or 7; and eight tumors had a Gleason score higher than 8. Sixty-seven percent (12 of 18) of the tumors were found to have perineural invasion, and 47% (9 of 19) had extracapsular invasion. A large observational cohort study, which included more than 6,000 MMR-variant carriers, reported an increased cumulative incidence of prostate cancer by age 70 years for specific MMR genes, as follows: MLH1 (7.0; 95% CI, 4.2–11.9), MSH2 (15.9; 95% CI, 11.2–22.5), and PMS2 (4.6; 95% CI, 0.8–67.5). No significant increase in prostate cancer incidence was reported for MSH6.[53]
Although the risk of prostate cancer appears to be elevated in families with Lynch syndrome, strategies for germline testing for MMR gene pathogenic variants in index prostate cancer patients remain to be determined.
A study of 1,133 primary prostate adenocarcinomas and 43 neuroendocrine prostate cancers (NEPC) conducted screening by MSH2 immunohistochemistry with confirmation by NGS.[54] MSI was assessed by polymerase chain reaction and NGS. Of primary adenocarcinomas and NEPC, 1.2% (14/1,176) had MSH2 loss. Overall, 8% (7/91) of adenocarcinomas with primary Gleason pattern 5 (Gleason score 9–10) had MSH2 loss compared with 0.4% (5/1,042) of tumors with any other Gleason scores (P < .05). Three patients had germline variants in MSH2, of whom two had a primary Gleason score of 5. Pending further confirmation, these findings may support universal MMR screening of prostate cancer with a Gleason score of 9 to 10 to identify men who may be eligible for immunotherapy and germline testing.
EPCAM testing has been included in some multigene panels likely due to EPCAM variants silencing MSH2. Specific large genomic rearrangement variants at the 3' end of EPCAM (which lies near the MSH2 gene) induce methylation of the MSH2 promoter, resulting in MSH2 protein loss.[55] Pathogenic variants in MSH2 are associated with Lynch syndrome and an increase in prostate cancer risk.[52] For more information on EPCAM and MSH2, see the Gene-specific considerations and associated CRC risk section or the Lynch Syndrome section in Genetics of Colorectal Cancer. Thus far, studies have not found an association between increased prostate cancer risk and EPCAM pathogenic variants.[56]
ATM
Ataxia telangiectasia (AT) is an autosomal recessive disorder characterized by neurological deterioration, telangiectasias, immunodeficiency states, and hypersensitivity to ionizing radiation. It is estimated that 1% of the general population may be heterozygous carriers of ATM pathogenic variants.[57] In the presence of DNA damage, the ATM protein is involved in mediating cell cycle arrest, DNA repair, and apoptosis.[58] Given evidence of other cancer risks in heterozygous ATM carriers, evidence of an association with prostate cancer susceptibility continues to emerge. A prospective case series of 10,317 Danish individuals who had a 36-year follow-up period, during which 2,056 individuals developed cancer, found that the ATM Ser49Cys variant was associated with increased prostate cancer risk (HR, 2.3; 95% CI, 1.1–5.0).[58] A retrospective case series of 692 men with metastatic prostate cancer, who were not selected based on a family history of cancer or the patient's age at cancer diagnosis, found that 1.6% of participants (11 of 692) had an ATM pathogenic variant.[56] Multiple independent reports have shown that the ATM P1054R variant, which is found in 2% of Europeans, is associated with increased prostate cancer risk.[37,59,60] For example, the Prostate Cancer Association Group to Investigate Cancer Associated Alterations in the Genome (PRACTICAL) consortium found an OR of 1.16 (95% CI, 1.10–1.22) for the ATM P1054 variant's association with prostate cancer risk.[61] A subsequent PRACTICAL consortium study had 14 groups (five from North America, six from Europe, and two from Australia) and 8,913 participants (5,560 cases and 3,353 controls). Next-generation ATM sequencing data were standardized and ClinVar classifications were used to categorize the variants as Tier 1 (likely pathogenic) or Tier 2 (potentially deleterious). Prostate cancer risk in Tier 1 variants had an OR of 4.4 (95% CI, 2.0–9.5).[62]
CHEK2
CHEK2 has also been investigated for a potential association with prostate cancer risk. For more information on other cancers associated with CHEK2 pathogenic variants, see the CHEK2 section in Genetics of Breast and Gynecologic Cancers and the CHEK2 section in Genetics of Colorectal Cancer. A retrospective case series of 692 men with metastatic prostate cancer unselected for cancer family history or age at diagnosis found 1.9% (10 of 534 [men with data]) were found to have a CHEK2 pathogenic variant.[56] A systematic review and meta-analysis from eight retrospective cohort studies examining the relationship between CHEK2 variants (1100delC, IVS2+1G>A, I157T) and prostate cancer confirmed the association of the 1100delC (OR, 3.29; 95% CI, 1.85–5.85; P = .00) and I157T (OR, 1.80; 95% CI, 1.51–2.14; P = .00) variants with prostate cancer susceptibility.[63] A genome-wide association study (GWAS) focusing on African American cases and controls identified a missense variant, I448S, which is associated with prostate cancer (risk allele frequency, 1.5%; OR, 1.62; 95% CI, 1.39–1.89, P = 7.50 × 10-10).[64] Further studies of CHEK2 in large diverse populations are warranted.
TP53
TP53 has also been investigated for a potential association with prostate cancer risk. For more information about other cancers associated with TP53 pathogenic variants, see the Li-Fraumeni Syndrome section in Genetics of Breast and Gynecologic Cancers. In a case series of 286 individuals from 107 families with a deleterious TP53 variant, 403 cancer diagnoses were reported, of which 211 were the first primary cancer including two prostate cancers diagnosed after age 45 years. Prostate cancer was also reported in 4 of 61 men with a second primary cancer.[65] In a Dutch case series of 180 families meeting either classic Li-Fraumeni syndrome (LFS) or Li-Fraumeni–like (LFL) family history criteria, a deleterious TP53 variant was identified in 24 families with one case of prostate cancer found in each group (LFS or LFL). Prostate cancer risks varied on the basis of the family history criteria with LFS (RR, 0.50; 95% CI, 0.01–3.00) and LFL (RR, 4.90; 95% CI, 0.10–27.00).[66] In a French case series of 415 families with a deleterious TP53 variant, four prostate cancers were reported, with a mean age at diagnosis of 63 years (range, 57–71 y).[67]
Germline TP53 pathogenic variants have also been identified in men with prostate cancer who have undergone tumor testing. A prospective case series of 42 men with either localized, biochemically recurrent, or metastatic prostate cancer unselected for cancer family history or age at diagnosis undergoing tumor-only somatic testing found that 2 of 42 men (5%) were found to have a suspected TP53 germline pathogenic variant.[68]
Further evidence supports an association between prostate cancer and germline TP53 pathogenic variants.[69,70,71] A retrospective study of 163 men (>18 y) with TP53 pathogenic/likely pathogenic variants from 132 known TP53 families found that 19% (n = 31/163) of participants had diagnoses of prostate cancer.[72] Of these participants, 48% (n = 31) were older than age 50 years. The median age of prostate cancer diagnosis was 56 years (range, 50–64 y). Locally advanced prostate cancer or de novo metastatic disease was found in 19% (n = 4) of men. Additionally, 40% (n = 8/20) of participants had high-grade prostate cancer (Gleason score, >8) at the time of diagnosis. This study also combined the existing cohort with a prostate cancer cohort that had documented germline TP53 pathogenic/likely pathogenic variants. This combined cohort had a prostate cancer relative risk of 9.1 (95% CI, 6.2–14; P < .0001).
NBN
NBN, which is also known as NBS1, has been investigated due to a potential association with prostate cancer risk, with the literature constantly evolving. Studies mostly from Polish populations reported that the NBN 657del5 variant is associated with prostate cancer risk (OR, 2.5; P < .001), mortality (HR, 1.6; P = .001), and familial prostate cancer (OR, 4.6; P < .0001).[73,74] One of these studies (from Poland) reported adverse survival when individuals with the NBN 657del5 variant also carried the NBN E185Q GG genotype (HR,1.9; P = .0004).[73] In the metastatic setting, a retrospective case series of 692 men with metastatic prostate cancer (unselected for cancer family history or age at diagnosis) found that 0.3% (2 of 692 men) had an NBN pathogenic variant.[56] Some clinical genetic testing laboratories do not include NBN on their prostate cancer panels, since NBN's association with prostate cancer is based on preliminary evidence. Further data will be required to fully understand the role and generalizability of NBN and its association with prostate cancer.
Multigene testing studies in prostate cancer
Prevalence of pathogenic variants with prostate cancer risk on multigene panel testing
The following section gives information about additional genes that may be on hereditary prostate cancer panel tests.
One retrospective case series of 692 men with metastatic prostate cancer unselected for cancer family history or age at diagnosis assessed the incidence of germline pathogenic variants in 16 DNA repair genes. Pathogenic variants were identified in 11.8% (82 of 692), a rate higher than in men with localized prostate cancer (4.6%, P < .001), suggesting that genetic aberrations are more commonly observed in men with aggressive forms of disease.[56] Two studies were published using data from a clinical testing laboratory database. The first study evaluated 1,328 men with prostate cancer and reported an overall pathogenic variant rate of 15.6%, including 10.9% in DNA repair genes.[75] A second study involved a larger cohort of 3,607 men with prostate cancer, some of whom had been included in the prior publication.[76] The reported pathogenic variant rate was 17.2%. Overall, pathogenic variant rates by gene were consistently reported between the two studies and were as follows: BRCA2, 4.74%; CHEK2, 2.88%; ATM, 2.03%; and BRCA1, 1.25%.[76] The most commonly aberrant gene in this cohort was BRCA2. The first publication reported associations between family history of breast cancer and high Gleason score (≥8).[75] The second publication focused on the percentage of men with pathogenic variants who met National Comprehensive Cancer Network national guidelines for genetic testing and found that 229 individuals (37%) with pathogenic variants in this cohort did not meet guidelines for genetic testing.[76] A systematic evidence review examined the median prevalence of pathogenic germline variants in the DNA damage-response pathway, including ATM, ATR, BRCA1, BRCA2, CHEK2, FANCA, MLH1, MRE11A, NBN, PALB2, and RAD51C. The overall prevalence was 18.6% (range, 17.2%–19%; n = 1,712) for general prostate cancer, 11.6% (range, 11.4%–11.8%; n = 1,261) for metastatic prostate cancer, 8.3% (range, 7.5%–9.1%; n = 738) for metastatic castration-resistant prostate cancer, and 29.3% (range, 7.3%–92.67%; n = 327) for familial prostate cancer.[77]
A case-control study in a Japanese population of 7,636 men with prostate cancer and 12,366 men without prostate cancer evaluated pathogenic variants in eight genes (BRCA1, BRCA2, CHEK2, ATM, NBN, PALB2, HOXB13, and BRIP1) for an association with prostate cancer.[37] The study found strong associations for BRCA2 (OR, 5.65; 95% CI, 3.55–9.32), HOXB13 (OR, 4.73; 95% CI, 2.84–8.19), and ATM (OR, 2.86; 95% CI, 1.63–5.15). The study supports a population-specific assessment of the genetic contribution to prostate cancer risk.
Germline pathogenic variants associated with metastatic prostate cancer
The metastatic prostate cancer setting is also contributing insights into the germline pathogenic variant spectrum of prostate cancer. Clinical sequencing of 150 metastatic tumors from men with castrate-resistant prostate cancer identified alterations in genes involved in DNA repair in 23% of men.[78] Interestingly, 8% of these variants were pathogenic and present in the germline. Another study focused on tumor-normal sequencing of advanced and metastatic cancers identified germline pathogenic variants in 19.6% of men (71 of 362) with prostate cancer.[79] Germline pathogenic variants were found in BRCA1, BRCA2, MSH2, MSH6, PALB2, PMS2, ATM, BRIP1, NBN, as well as other genes. These and other studies are summarized in Table 6. The contribution of germline variants identified from large sequencing efforts to inherited prostate cancer predisposition requires molecular confirmation of genes not classically linked to prostate cancer risk.
| Study | Cohort | Germline Results for Prostate Cancer | Comments | ||
|---|---|---|---|---|---|
| mCRPC = metastatic castration-resistant prostate cancer. | |||||
| a Potential overlap of cohorts. | |||||
| Robinson et al. (2015)a[78] | Whole-exomeand transcriptome sequencing of bone or soft tissue tumor biopsies from a cohort of 150 men with mCRPC | 8% had germline pathogenic variants: | |||
| —BRCA2: 9/150 (6.0%) | |||||
| —ATM: 2/150 (1.3%) | |||||
| —BRCA1: 1/150 (0.7%) | |||||
| Pritchard et al. (2016)a[56] | 692 men with metastatic prostate cancer, unselected for family history; analysis focused on 20 genes involved in maintaining DNA integrity and associated withautosomal dominantcancer–predisposing syndromes | 82/692 (11.8%) had germline pathogenic variants: | Frequency of germline pathogenic variants in DNA repair genes among men with metastatic prostate cancer significantly exceeded the prevalence of 4.6% among 499 men with localized prostate cancer in the Cancer Genome Atlas (P < .001) | ||
| —BRCA2: 37/692 (5.3%) | |||||
| —ATM: 11/692 (1.6%) | |||||
| —BRCA1: 6/692 (0.9%) | |||||
| Schrader et al. (2016)[80] | 1,566 patients undergoing tumor profiling (341 genes) with matched normal DNA at a single institution; 97 cases of prostate cancer included | 10/97 (10.3%) had germline pathogenic variants: | |||
| —BRCA2: 6/97 (6.2%) | |||||
| —BRCA1: 1/97 (1.0%) | |||||
| —MSH6: 1/97 (1.0%) | |||||
| —MUTYH: 1/97 (1.0%) | |||||
| —PMS2: 1/97 (1.0%) | |||||
Common Risk Variants and Polygenic Risk Scores for Prostate Cancer
The most prevalent prostate cancer risk variants in the human genome were discovered in genome-wide association studies (GWAS). GWAS evaluate the millions of common single nucleotide polymorphisms (SNPs) in the human population (typically >5% prevalence) and ask if each variant is enriched in individuals with a given disease. With great statistical rigor, GWAS have revealed over 250 prostate cancer risk variants. Each single SNP confers a very modest prostate cancer risk. However, when compounded, these SNPs comprise a substantial portion of inherited prostate cancer risk. Research continues to translate these discoveries into clinical practice, with use in tools like polygenic risk scores (PRS).
GWAS and SNPs
- GWAS can identify inherited genetic variants that influence a specific phenotype, such as risk of a particular disease.
- For complex diseases, such as prostate cancer, risk of developing the disease is the product of multiple genetic and environmental factors; each individual factor contributes relatively little to overall risk.
- To date, GWAS have discovered more than 250 common genetic variants associated with prostate cancer risk.
- Individuals can be genotyped for all known prostate cancer risk markers relatively easily; but, to date, studies have not demonstrated that this information substantially refines risk estimates from commonly used variables, such as family history.
- The clinical relevance of variants identified from GWAS remains unclear.
Although the statistical evidence for an association between genetic variation at these loci and prostate cancer risk is overwhelming, the clinical relevance of the variants and the mechanism(s) by which they lead to increased risk are unclear and will require further characterization. Additionally, these loci are associated with very modest risk estimates and explain only a fraction of overall inherited risk. However, when combined into a PRS, these confirmed genetic risk variants may prove to be useful for prostate cancer risk stratification and to identify men for targeted screening and early detection. Further work will include genome-wide analysis of rarer alleles catalogued via sequencing efforts. Disease-associated alleles with frequencies of less than 1% in the population may prove to be more highly penetrant and clinically useful. In addition, further work is needed to describe the landscape of genetic risk in non-European populations. Finally, until the individual and collective influences of genetic risk alleles are evaluated prospectively, their clinical utility will remain difficult to fully assess.
Beginning in 2006, multiple genome-wide studies seeking associations with prostate cancer risk converged on the same chromosomal locus, 8q24.[81,82,83,84,85,86,87,88,89,90,91,92,93,94] Since that time, more than ten genetic polymorphisms, all independently associated with disease, reside within five distinct 8q24 risk regions. The population-attributable risk of prostate cancer from the 8q24 risk alleles reported to date is 9.4%.[95]
Since prostate cancer risk loci have been discovered at 8q24, more than 250 variants have been identified at other chromosomal risk loci. These chromosomal risk loci were detected by multistage GWAS, which were comprised of thousands of cases and controls and were validated in independent cohorts.[96] The most convincing associations reported to date for men of European ancestry are annotated in the National Human Genome Research Institute GWAS catalog.
Most prostate cancer GWAS data generated to date have been derived from populations of European descent. This shortcoming is profound, considering that linkage disequilibrium structure, SNV frequencies, and incidence of disease differ across ancestral groups. To provide meaningful genetic data to all patients, well-designed, adequately powered GWAS must be aimed at specific ethnic groups.[97] Most work in this regard has focused on African American, Chinese, and Japanese men. The most convincing associations reported to date for men of non-European ancestry are annotated in the National Human Genome Research Institute GWAS catalog.
The African American population is of particular interest because American men with West African ancestry are at higher risk of prostate cancer than any other group. A handful of studies have sought to determine whether GWAS findings in men of European ancestry are applicable to men of African ancestry.[64,98,99] The majority of risk alleles (approximately 83%) are shared across African American and European American populations. Three independent associations were subsequently replicated. All three variants were within or near long noncoding RNAs (lncRNAs) previously associated with prostate cancer, and two of the variants were unique to men of African ancestry.[100]
Statistically well-powered GWAS have also been launched to examine inherited cancer risk in Japanese and Chinese populations. Investigators discovered that these populations share many risk regions observed in African American men.[101,102,103,104] Additionally, risk regions that are unique to these ancestral groups were identified (for more information, see the National Human Genome Research Institute GWAS catalog). Ongoing work in larger cohorts will validate and expand upon these findings.
Polygenic risk scores for prostate cancer
Current GWAS findings account for an estimated 58% of heritable prostate cancer risk. Another 6% of familial prostate cancer risk is attributed to rare genetic variants.[105] Efforts have been made to translate these discoveries into clinically useful metrics for risk stratification and early detection. PRS were devised to measure prostate cancer risk based on the burden of genetic risk variants that an individual inherits. Associations between PRS and disease risk clearly exist. However, it remains unclear whether screening PRS can appreciably influence long-term outcomes.
In a 2023 study, PRS were created for a multi-ethnic cohort of over 150,000 prostate cancer cases and over 750,000 controls.[106] A PRS was based on 451 prostate cancer risk variants validated via GWAS. The study accounted for genetic dose (i.e., homozygosity vs. heterozygosity). When focusing on men in the top quintile of PRS scores and comparing them to men in the middle of the distribution, men of European ancestry had an OR of greater than 2-fold for developing prostate cancer when compared with men who had average PRS scores. In men of African ancestry, those who belonged to the upper 16% of the PRS had a greater than 2-fold increased risk to develop prostate cancer before age 66 years when compared with those who had average PRS scores. Men in the upper quintile of the PRS represented over 50% of prostate cancer cases, including clinically aggressive cases. In contrast, those in the lowest quintile of the PRS represented fewer than 5% of prostate cancer cases. These data suggest that PRS could inform prostate cancer screening.[107,108] Studies that were conducted prior to this 2023 study analyzed multi-ethnic cohorts and began validating models.[109,110,111,112,113,114,115,116,117,118,119,120] Further research is needed to determine whether a PRS devised using prostate cancer risk SNPs can help identify clinically aggressive disease.[121]
As GWAS elucidate these networks, it is hoped that new therapies and chemopreventive strategies will follow.[122,123,124,125,126,127,128,129,130]
Germline SNPs associated with prostate cancer aggressiveness
Prostate cancer is biologically and clinically heterogeneous. Many tumors are indolent and are successfully managed with observation alone. Other tumors are quite aggressive and prove deadly. Several variables are used to determine prostate cancer aggressiveness at the time of diagnosis, such as Gleason score and PSA, but these are imperfect. Additional markers are needed because sound treatment decisions depend on accurate prognostic information. Germline genetic variants are attractive markers because they are present, easily detectable, and static throughout life.
Findings regarding inherited risk of aggressive disease are considered preliminary. Further work is needed to validate findings and assess these associations prospectively.
References:
- Mucci LA, Hjelmborg JB, Harris JR, et al.: Familial Risk and Heritability of Cancer Among Twins in Nordic Countries. JAMA 315 (1): 68-76, 2016.
- Thompson D, Easton DF; Breast Cancer Linkage Consortium: Cancer Incidence in BRCA1 mutation carriers. J Natl Cancer Inst 94 (18): 1358-65, 2002.
- Nyberg T, Tischkowitz M, Antoniou AC: BRCA1 and BRCA2 pathogenic variants and prostate cancer risk: systematic review and meta-analysis. Br J Cancer 126 (7): 1067-1081, 2022.
- Mersch J, Jackson MA, Park M, et al.: Cancers associated with BRCA1 and BRCA2 mutations other than breast and ovarian. Cancer 121 (2): 269-75, 2015.
- Nastiuk KL, Mansukhani M, Terry MB, et al.: Common mutations in BRCA1 and BRCA2 do not contribute to early prostate cancer in Jewish men. Prostate 40 (3): 172-7, 1999.
- Vazina A, Baniel J, Yaacobi Y, et al.: The rate of the founder Jewish mutations in BRCA1 and BRCA2 in prostate cancer patients in Israel. Br J Cancer 83 (4): 463-6, 2000.
- Lehrer S, Fodor F, Stock RG, et al.: Absence of 185delAG mutation of the BRCA1 gene and 6174delT mutation of the BRCA2 gene in Ashkenazi Jewish men with prostate cancer. Br J Cancer 78 (6): 771-3, 1998.
- Struewing JP, Abeliovich D, Peretz T, et al.: The carrier frequency of the BRCA1 185delAG mutation is approximately 1 percent in Ashkenazi Jewish individuals. Nat Genet 11 (2): 198-200, 1995.
- Oddoux C, Struewing JP, Clayton CM, et al.: The carrier frequency of the BRCA2 6174delT mutation among Ashkenazi Jewish individuals is approximately 1%. Nat Genet 14 (2): 188-90, 1996.
- Roa BB, Boyd AA, Volcik K, et al.: Ashkenazi Jewish population frequencies for common mutations in BRCA1 and BRCA2. Nat Genet 14 (2): 185-7, 1996.
- Struewing JP, Hartge P, Wacholder S, et al.: The risk of cancer associated with specific mutations of BRCA1 and BRCA2 among Ashkenazi Jews. N Engl J Med 336 (20): 1401-8, 1997.
- Nyberg T, Frost D, Barrowdale D, et al.: Prostate Cancer Risks for Male BRCA1 and BRCA2 Mutation Carriers: A Prospective Cohort Study. Eur Urol 77 (1): 24-35, 2020.
- Patel VL, Busch EL, Friebel TM, et al.: Association of Genomic Domains in BRCA1 and BRCA2 with Prostate Cancer Risk and Aggressiveness. Cancer Res 80 (3): 624-638, 2020.
- National Comprehensive Cancer Network: NCCN Clinical Practice Guidelines in Oncology: Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic. Version 2.2024. Plymouth Meeting, Pa: National Comprehensive Cancer Network, 2023. Available online with free registration. Last accessed October 31, 2023.
- Taylor RA, Fraser M, Livingstone J, et al.: Germline BRCA2 mutations drive prostate cancers with distinct evolutionary trajectories. Nat Commun 8: 13671, 2017.
- Oh M, Alkhushaym N, Fallatah S, et al.: The association of BRCA1 and BRCA2 mutations with prostate cancer risk, frequency, and mortality: A meta-analysis. Prostate 79 (8): 880-895, 2019.
- Lange EM, Gillanders EM, Davis CC, et al.: Genome-wide scan for prostate cancer susceptibility genes using families from the University of Michigan prostate cancer genetics project finds evidence for linkage on chromosome 17 near BRCA1. Prostate 57 (4): 326-34, 2003.
- Lange EM, Robbins CM, Gillanders EM, et al.: Fine-mapping the putative chromosome 17q21-22 prostate cancer susceptibility gene to a 10 cM region based on linkage analysis. Hum Genet 121 (1): 49-55, 2007.
- Ewing CM, Ray AM, Lange EM, et al.: Germline mutations in HOXB13 and prostate-cancer risk. N Engl J Med 366 (2): 141-9, 2012.
- Schroeck FR, Zuhlke KA, Siddiqui J, et al.: Testing for the recurrent HOXB13 G84E germline mutation in men with clinical indications for prostate biopsy. J Urol 189 (3): 849-53, 2013.
- National Comprehensive Cancer Network: NCCN Clinical Practice Guidelines in Oncology: Prostate Cancer Early Detection. Version 2.2023. Plymouth Meeting, Pa: National Comprehensive Cancer Network, 2023. Available online with free registration. Last accessed November 30, 2023.
- Xu J, Lange EM, Lu L, et al.: HOXB13 is a susceptibility gene for prostate cancer: results from the International Consortium for Prostate Cancer Genetics (ICPCG). Hum Genet 132 (1): 5-14, 2013.
- Chen Z, Greenwood C, Isaacs WB, et al.: The G84E mutation of HOXB13 is associated with increased risk for prostate cancer: results from the REDUCE trial. Carcinogenesis 34 (6): 1260-4, 2013.
- Shang Z, Zhu S, Zhang H, et al.: Germline homeobox B13 (HOXB13) G84E mutation and prostate cancer risk in European descendants: a meta-analysis of 24,213 cases and 73, 631 controls. Eur Urol 64 (1): 173-6, 2013.
- Handorf E, Crumpler N, Gross L, et al.: Prevalence of the HOXB13 G84E mutation among unaffected men with a family history of prostate cancer. J Genet Couns 23 (3): 371-6, 2014.
- Laitinen VH, Wahlfors T, Saaristo L, et al.: HOXB13 G84E mutation in Finland: population-based analysis of prostate, breast, and colorectal cancer risk. Cancer Epidemiol Biomarkers Prev 22 (3): 452-60, 2013.
- Witte JS, Mefford J, Plummer SJ, et al.: HOXB13 mutation and prostate cancer: studies of siblings and aggressive disease. Cancer Epidemiol Biomarkers Prev 22 (4): 675-80, 2013.
- Beebe-Dimmer JL, Hathcock M, Yee C, et al.: The HOXB13 G84E Mutation Is Associated with an Increased Risk for Prostate Cancer and Other Malignancies. Cancer Epidemiol Biomarkers Prev 24 (9): 1366-72, 2015.
- Gudmundsson J, Sulem P, Gudbjartsson DF, et al.: A study based on whole-genome sequencing yields a rare variant at 8q24 associated with prostate cancer. Nat Genet 44 (12): 1326-9, 2012.
- Huang H, Cai B: G84E mutation in HOXB13 is firmly associated with prostate cancer risk: a meta-analysis. Tumour Biol 35 (2): 1177-82, 2014.
- Cai Q, Wang X, Li X, et al.: Germline HOXB13 p.Gly84Glu mutation and cancer susceptibility: a pooled analysis of 25 epidemiological studies with 145,257 participates. Oncotarget 6 (39): 42312-21, 2015.
- Stott-Miller M, Karyadi DM, Smith T, et al.: HOXB13 mutations in a population-based, case-control study of prostate cancer. Prostate 73 (6): 634-41, 2013.
- Alanee S, Shah S, Vijai J, et al.: Prevalence of HOXB13 mutation in a population of Ashkenazi Jewish men treated for prostate cancer. Fam Cancer 12 (4): 597-600, 2013.
- Kote-Jarai Z, Mikropoulos C, Leongamornlert DA, et al.: Prevalence of the HOXB13 G84E germline mutation in British men and correlation with prostate cancer risk, tumour characteristics and clinical outcomes. Ann Oncol 26 (4): 756-61, 2015.
- Sipeky C, Gao P, Zhang Q, et al.: Synergistic Interaction of HOXB13 and CIP2A Predisposes to Aggressive Prostate Cancer. Clin Cancer Res 24 (24): 6265-6276, 2018.
- Lin X, Qu L, Chen Z, et al.: A novel germline mutation in HOXB13 is associated with prostate cancer risk in Chinese men. Prostate 73 (2): 169-75, 2013.
- Momozawa Y, Iwasaki Y, Hirata M, et al.: Germline Pathogenic Variants in 7636 Japanese Patients With Prostate Cancer and 12 366 Controls. J Natl Cancer Inst 112 (4): 369-376, 2020.
- Marlin R, Créoff M, Merle S, et al.: Mutation HOXB13 c.853delT in Martinican prostate cancer patients. Prostate 80 (6): 463-470, 2020.
- Na R, Wei J, Sample CJ, et al.: The HOXB13 variant X285K is associated with clinical significance and early age at diagnosis in African American prostate cancer patients. Br J Cancer 126 (5): 791-796, 2022.
- Darst BF, Hughley R, Pfennig A, et al.: A Rare Germline HOXB13 Variant Contributes to Risk of Prostate Cancer in Men of African Ancestry. Eur Urol 81 (5): 458-462, 2022.
- Karlsson R, Aly M, Clements M, et al.: A population-based assessment of germline HOXB13 G84E mutation and prostate cancer risk. Eur Urol 65 (1): 169-76, 2014.
- MacInnis RJ, Severi G, Baglietto L, et al.: Population-based estimate of prostate cancer risk for carriers of the HOXB13 missense mutation G84E. PLoS One 8 (2): e54727, 2013.
- Nyberg T, Govindasami K, Leslie G, et al.: Homeobox B13 G84E Mutation and Prostate Cancer Risk. Eur Urol 75 (5): 834-845, 2019.
- Soravia C, van der Klift H, Bründler MA, et al.: Prostate cancer is part of the hereditary non-polyposis colorectal cancer (HNPCC) tumor spectrum. Am J Med Genet 121A (2): 159-62, 2003.
- Haraldsdottir S, Hampel H, Wei L, et al.: Prostate cancer incidence in males with Lynch syndrome. Genet Med 16 (7): 553-7, 2014.
- Grindedal EM, Møller P, Eeles R, et al.: Germ-line mutations in mismatch repair genes associated with prostate cancer. Cancer Epidemiol Biomarkers Prev 18 (9): 2460-7, 2009.
- Langeberg WJ, Kwon EM, Koopmeiners JS, et al.: Population-based study of the association of variants in mismatch repair genes with prostate cancer risk and outcomes. Cancer Epidemiol Biomarkers Prev 19 (1): 258-64, 2010.
- Bauer CM, Ray AM, Halstead-Nussloch BA, et al.: Hereditary prostate cancer as a feature of Lynch syndrome. Fam Cancer 10 (1): 37-42, 2011.
- Dominguez-Valentin M, Joost P, Therkildsen C, et al.: Frequent mismatch-repair defects link prostate cancer to Lynch syndrome. BMC Urol 16: 15, 2016.
- Raymond VM, Mukherjee B, Wang F, et al.: Elevated risk of prostate cancer among men with Lynch syndrome. J Clin Oncol 31 (14): 1713-8, 2013.
- Ryan S, Jenkins MA, Win AK: Risk of prostate cancer in Lynch syndrome: a systematic review and meta-analysis. Cancer Epidemiol Biomarkers Prev 23 (3): 437-49, 2014.
- Rosty C, Walsh MD, Lindor NM, et al.: High prevalence of mismatch repair deficiency in prostate cancers diagnosed in mismatch repair gene mutation carriers from the colon cancer family registry. Fam Cancer 13 (4): 573-82, 2014.
- Dominguez-Valentin M, Sampson JR, Seppälä TT, et al.: Cancer risks by gene, age, and gender in 6350 carriers of pathogenic mismatch repair variants: findings from the Prospective Lynch Syndrome Database. Genet Med 22 (1): 15-25, 2020.
- Guedes LB, Antonarakis ES, Schweizer MT, et al.: MSH2 Loss in Primary Prostate Cancer. Clin Cancer Res 23 (22): 6863-6874, 2017.
- Kovacs ME, Papp J, Szentirmay Z, et al.: Deletions removing the last exon of TACSTD1 constitute a distinct class of mutations predisposing to Lynch syndrome. Hum Mutat 30 (2): 197-203, 2009.
- Pritchard CC, Mateo J, Walsh MF, et al.: Inherited DNA-Repair Gene Mutations in Men with Metastatic Prostate Cancer. N Engl J Med 375 (5): 443-53, 2016.
- Savitsky K, Bar-Shira A, Gilad S, et al.: A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science 268 (5218): 1749-53, 1995.
- Dombernowsky SL, Weischer M, Allin KH, et al.: Risk of cancer by ATM missense mutations in the general population. J Clin Oncol 26 (18): 3057-62, 2008.
- Angèle S, Falconer A, Edwards SM, et al.: ATM polymorphisms as risk factors for prostate cancer development. Br J Cancer 91 (4): 783-7, 2004.
- Meyer A, Wilhelm B, Dörk T, et al.: ATM missense variant P1054R predisposes to prostate cancer. Radiother Oncol 83 (3): 283-8, 2007.
- Schumacher FR, Al Olama AA, Berndt SI, et al.: Association analyses of more than 140,000 men identify 63 new prostate cancer susceptibility loci. Nat Genet 50 (7): 928-936, 2018.
- Karlsson Q, Brook MN, Dadaev T, et al.: Rare Germline Variants in ATM Predispose to Prostate Cancer: A PRACTICAL Consortium Study. Eur Urol Oncol 4 (4): 570-579, 2021.
- Wang Y, Dai B, Ye D: CHEK2 mutation and risk of prostate cancer: a systematic review and meta-analysis. Int J Clin Exp Med 8 (9): 15708-15, 2015.
- Conti DV, Wang K, Sheng X, et al.: Two Novel Susceptibility Loci for Prostate Cancer in Men of African Ancestry. J Natl Cancer Inst 109 (8): , 2017.
- Mai PL, Best AF, Peters JA, et al.: Risks of first and subsequent cancers among TP53 mutation carriers in the National Cancer Institute Li-Fraumeni syndrome cohort. Cancer 122 (23): 3673-3681, 2016.
- Ruijs MW, Verhoef S, Rookus MA, et al.: TP53 germline mutation testing in 180 families suspected of Li-Fraumeni syndrome: mutation detection rate and relative frequency of cancers in different familial phenotypes. J Med Genet 47 (6): 421-8, 2010.
- Bougeard G, Renaux-Petel M, Flaman JM, et al.: Revisiting Li-Fraumeni Syndrome From TP53 Mutation Carriers. J Clin Oncol 33 (21): 2345-52, 2015.
- Cheng HH, Klemfuss N, Montgomery B, et al.: A Pilot Study of Clinical Targeted Next Generation Sequencing for Prostate Cancer: Consequences for Treatment and Genetic Counseling. Prostate 76 (14): 1303-11, 2016.
- Stacey SN, Sulem P, Jonasdottir A, et al.: A germline variant in the TP53 polyadenylation signal confers cancer susceptibility. Nat Genet 43 (11): 1098-103, 2011.
- Mittal RD, George GP, Mishra J, et al.: Role of functional polymorphisms of P53 and P73 genes with the risk of prostate cancer in a case-control study from Northern India. Arch Med Res 42 (2): 122-7, 2011.
- Xu B, Xu Z, Cheng G, et al.: Association between polymorphisms of TP53 and MDM2 and prostate cancer risk in southern Chinese. Cancer Genet Cytogenet 202 (2): 76-81, 2010.
- Maxwell KN, Cheng HH, Powers J, et al.: Inherited TP53 Variants and Risk of Prostate Cancer. Eur Urol 81 (3): 243-250, 2022.
- Rusak B, Kluźniak W, Wokołorczykv D, et al.: Inherited NBN Mutations and Prostate Cancer Risk and Survival. Cancer Res Treat 51 (3): 1180-1187, 2019.
- Wokołorczyk D, Kluźniak W, Huzarski T, et al.: Mutations in ATM, NBN and BRCA2 predispose to aggressive prostate cancer in Poland. Int J Cancer 147 (10): 2793-2800, 2020.
- Giri VN, Hegarty SE, Hyatt C, et al.: Germline genetic testing for inherited prostate cancer in practice: Implications for genetic testing, precision therapy, and cascade testing. Prostate 79 (4): 333-339, 2019.
- Nicolosi P, Ledet E, Yang S, et al.: Prevalence of Germline Variants in Prostate Cancer and Implications for Current Genetic Testing Guidelines. JAMA Oncol 5 (4): 523-528, 2019.
- Lang SH, Swift SL, White H, et al.: A systematic review of the prevalence of DNA damage response gene mutations in prostate cancer. Int J Oncol 55 (3): 597-616, 2019.
- Robinson D, Van Allen EM, Wu YM, et al.: Integrative clinical genomics of advanced prostate cancer. Cell 161 (5): 1215-28, 2015.
- Mandelker D, Zhang L, Kemel Y, et al.: Mutation Detection in Patients With Advanced Cancer by Universal Sequencing of Cancer-Related Genes in Tumor and Normal DNA vs Guideline-Based Germline Testing. JAMA 318 (9): 825-835, 2017.
- Schrader KA, Cheng DT, Joseph V, et al.: Germline Variants in Targeted Tumor Sequencing Using Matched Normal DNA. JAMA Oncol 2 (1): 104-11, 2016.
- Amundadottir LT, Sulem P, Gudmundsson J, et al.: A common variant associated with prostate cancer in European and African populations. Nat Genet 38 (6): 652-8, 2006.
- Schumacher FR, Feigelson HS, Cox DG, et al.: A common 8q24 variant in prostate and breast cancer from a large nested case-control study. Cancer Res 67 (7): 2951-6, 2007.
- Suuriniemi M, Agalliu I, Schaid DJ, et al.: Confirmation of a positive association between prostate cancer risk and a locus at chromosome 8q24. Cancer Epidemiol Biomarkers Prev 16 (4): 809-14, 2007.
- Wang L, McDonnell SK, Slusser JP, et al.: Two common chromosome 8q24 variants are associated with increased risk for prostate cancer. Cancer Res 67 (7): 2944-50, 2007.
- Yeager M, Orr N, Hayes RB, et al.: Genome-wide association study of prostate cancer identifies a second risk locus at 8q24. Nat Genet 39 (5): 645-9, 2007.
- Zheng SL, Sun J, Cheng Y, et al.: Association between two unlinked loci at 8q24 and prostate cancer risk among European Americans. J Natl Cancer Inst 99 (20): 1525-33, 2007.
- Savage SA, Greene MH: The evidence for prostate cancer risk loci at 8q24 grows stronger. J Natl Cancer Inst 99 (20): 1499-501, 2007.
- Salinas CA, Kwon E, Carlson CS, et al.: Multiple independent genetic variants in the 8q24 region are associated with prostate cancer risk. Cancer Epidemiol Biomarkers Prev 17 (5): 1203-13, 2008.
- Zheng SL, Hsing AW, Sun J, et al.: Association of 17 prostate cancer susceptibility loci with prostate cancer risk in Chinese men. Prostate 70 (4): 425-32, 2010.
- Zeegers MP, Khan HS, Schouten LJ, et al.: Genetic marker polymorphisms on chromosome 8q24 and prostate cancer in the Dutch population: DG8S737 may not be the causative variant. Eur J Hum Genet 19 (1): 118-20, 2011.
- Gudmundsson J, Sulem P, Manolescu A, et al.: Genome-wide association study identifies a second prostate cancer susceptibility variant at 8q24. Nat Genet 39 (5): 631-7, 2007.
- Haiman CA, Patterson N, Freedman ML, et al.: Multiple regions within 8q24 independently affect risk for prostate cancer. Nat Genet 39 (5): 638-44, 2007.
- Yeager M, Chatterjee N, Ciampa J, et al.: Identification of a new prostate cancer susceptibility locus on chromosome 8q24. Nat Genet 41 (10): 1055-7, 2009.
- Al Olama AA, Kote-Jarai Z, Giles GG, et al.: Multiple loci on 8q24 associated with prostate cancer susceptibility. Nat Genet 41 (10): 1058-60, 2009.
- Matejcic M, Saunders EJ, Dadaev T, et al.: Germline variation at 8q24 and prostate cancer risk in men of European ancestry. Nat Commun 9 (1): 4616, 2018.
- Conti DV, Darst BF, Moss LC, et al.: Trans-ancestry genome-wide association meta-analysis of prostate cancer identifies new susceptibility loci and informs genetic risk prediction. Nat Genet 53 (1): 65-75, 2021.
- Cook MB, Wang Z, Yeboah ED, et al.: A genome-wide association study of prostate cancer in West African men. Hum Genet 133 (5): 509-21, 2014.
- Haiman CA, Chen GK, Blot WJ, et al.: Characterizing genetic risk at known prostate cancer susceptibility loci in African Americans. PLoS Genet 7 (5): e1001387, 2011.
- Han Y, Signorello LB, Strom SS, et al.: Generalizability of established prostate cancer risk variants in men of African ancestry. Int J Cancer 136 (5): 1210-7, 2015.
- Han Y, Rand KA, Hazelett DJ, et al.: Prostate Cancer Susceptibility in Men of African Ancestry at 8q24. J Natl Cancer Inst 108 (7): , 2016.
- Takata R, Akamatsu S, Kubo M, et al.: Genome-wide association study identifies five new susceptibility loci for prostate cancer in the Japanese population. Nat Genet 42 (9): 751-4, 2010.
- Akamatsu S, Takata R, Haiman CA, et al.: Common variants at 11q12, 10q26 and 3p11.2 are associated with prostate cancer susceptibility in Japanese. Nat Genet 44 (4): 426-9, S1, 2012.
- Xu J, Mo Z, Ye D, et al.: Genome-wide association study in Chinese men identifies two new prostate cancer risk loci at 9q31.2 and 19q13.4. Nat Genet 44 (11): 1231-5, 2012.
- Takata R, Takahashi A, Fujita M, et al.: 12 new susceptibility loci for prostate cancer identified by genome-wide association study in Japanese population. Nat Commun 10 (1): 4422, 2019.
- Benafif S, Kote-Jarai Z, Eeles RA, et al.: A Review of Prostate Cancer Genome-Wide Association Studies (GWAS). Cancer Epidemiol Biomarkers Prev 27 (8): 845-857, 2018.
- Wang A, Shen J, Rodriguez AA, et al.: Characterizing prostate cancer risk through multi-ancestry genome-wide discovery of 187 novel risk variants. Nat Genet 55 (12): 2065-2074, 2023.
- Chou A, Darst BF, Wilkens LR, et al.: Association of Prostate-Specific Antigen Levels with Prostate Cancer Risk in a Multiethnic Population: Stability Over Time and Comparison with Polygenic Risk Score. Cancer Epidemiol Biomarkers Prev 31 (12): 2199-2207, 2022.
- Kachuri L, Hoffmann TJ, Jiang Y, et al.: Genetically adjusted PSA levels for prostate cancer screening. Nat Med 29 (6): 1412-1423, 2023.
- Nyberg T, Brook MN, Ficorella L, et al.: CanRisk-Prostate: A Comprehensive, Externally Validated Risk Model for the Prediction of Future Prostate Cancer. J Clin Oncol 41 (5): 1092-1104, 2023.
- Dite GS, Spaeth E, Murphy NM, et al.: Development and validation of a simple prostate cancer risk prediction model based on age, family history, and polygenic risk. Prostate 83 (10): 962-969, 2023.
- Black MH, Li S, LaDuca H, et al.: Validation of a prostate cancer polygenic risk score. Prostate 80 (15): 1314-1321, 2020.
- Yoon BW, Shin HT, Seo JH: Risk Allele Frequency Analysis and Risk Prediction of Single-Nucleotide Polymorphisms for Prostate Cancer. Genes (Basel) 13 (11): , 2022.
- Huntley C, Torr B, Sud A, et al.: Utility of polygenic risk scores in UK cancer screening: a modelling analysis. Lancet Oncol 24 (6): 658-668, 2023.
- Karunamuni RA, Huynh-Le MP, Fan CC, et al.: Performance of African-ancestry-specific polygenic hazard score varies according to local ancestry in 8q24. Prostate Cancer Prostatic Dis 25 (2): 229-237, 2022.
- Chen F, Madduri RK, Rodriguez AA, et al.: Evidence of Novel Susceptibility Variants for Prostate Cancer and a Multiancestry Polygenic Risk Score Associated with Aggressive Disease in Men of African Ancestry. Eur Urol 84 (1): 13-21, 2023.
- Zhang W, Nicholson T, Zhang K: Deciphering the Polygenic Basis of Racial Disparities in Prostate Cancer By an Integrative Analysis of Genomic and Transcriptomic Data. Cancer Prev Res (Phila) 15 (3): 161-171, 2022.
- Ruan X, Huang D, Huang J, et al.: Application of European-specific polygenic risk scores for predicting prostate cancer risk in different ancestry populations. Prostate 83 (1): 30-38, 2023.
- Darst BF, Shen J, Madduri RK, et al.: Evaluating approaches for constructing polygenic risk scores for prostate cancer in men of African and European ancestry. Am J Hum Genet 110 (7): 1200-1206, 2023.
- Siltari A, Lönnerbro R, Pang K, et al.: How Well do Polygenic Risk Scores Identify Men at High Risk for Prostate Cancer? Systematic Review and Meta-Analysis. Clin Genitourin Cancer 21 (2): 316.e1-316.e11, 2023.
- Chen F, Darst BF, Madduri RK, et al.: Validation of a multi-ancestry polygenic risk score and age-specific risks of prostate cancer: A meta-analysis within diverse populations. Elife 11: , 2022.
- Ruan X, Huang D, Huang J, et al.: Genetic risk assessment of lethal prostate cancer using polygenic risk score and hereditary cancer susceptibility genes. J Transl Med 21 (1): 446, 2023.
- Freedman ML, Monteiro AN, Gayther SA, et al.: Principles for the post-GWAS functional characterization of cancer risk loci. Nat Genet 43 (6): 513-8, 2011.
- Pomerantz MM, Beckwith CA, Regan MM, et al.: Evaluation of the 8q24 prostate cancer risk locus and MYC expression. Cancer Res 69 (13): 5568-74, 2009.
- Jia L, Landan G, Pomerantz M, et al.: Functional enhancers at the gene-poor 8q24 cancer-linked locus. PLoS Genet 5 (8): e1000597, 2009.
- Ahmadiyeh N, Pomerantz MM, Grisanzio C, et al.: 8q24 prostate, breast, and colon cancer risk loci show tissue-specific long-range interaction with MYC. Proc Natl Acad Sci U S A 107 (21): 9742-6, 2010.
- Sotelo J, Esposito D, Duhagon MA, et al.: Long-range enhancers on 8q24 regulate c-Myc. Proc Natl Acad Sci U S A 107 (7): 3001-5, 2010.
- Meyer KB, Maia AT, O'Reilly M, et al.: A functional variant at a prostate cancer predisposition locus at 8q24 is associated with PVT1 expression. PLoS Genet 7 (7): e1002165, 2011.
- Spisák S, Lawrenson K, Fu Y, et al.: CAUSEL: an epigenome- and genome-editing pipeline for establishing function of noncoding GWAS variants. Nat Med 21 (11): 1357-63, 2015.
- Hazelett DJ, Rhie SK, Gaddis M, et al.: Comprehensive functional annotation of 77 prostate cancer risk loci. PLoS Genet 10 (1): e1004102, 2014.
- Jiang J, Cui W, Vongsangnak W, et al.: Post genome-wide association studies functional characterization of prostate cancer risk loci. BMC Genomics 14 (Suppl 8): S9, 2013.
This section addresses the impact of genetics on prostate cancer screening, surveillance, and treatment. For more information about prostate cancer screening, surveillance, and treatment, see Prostate Cancer Screening and Prostate Cancer Treatment.
Prostate Cancer Screening
Background
Decisions about risk-reducing interventions for patients with an inherited predisposition to prostate cancer, as with any disease, are best guided by randomized controlled clinical trials and knowledge of the underlying natural history of the process. However, existing studies of screening for prostate cancer in high-risk men (men with a positive family history of prostate cancer and African American men) are predominantly based on retrospective case series or retrospective cohort analyses. Because awareness of a positive family history can lead to more frequent work-ups for cancer and result in apparently earlier prostate cancer detection, assessments of disease progression rates and survival after diagnosis are subject to selection, lead time, and length biases. This section focuses on screening and risk reduction of prostate cancer among men predisposed to the disease; data relevant to screening in high-risk men are primarily extracted from studies performed in the general population.
Screening
Information is limited about the efficacy of commonly available screening tests such as the digital rectal exam (DRE) and serum prostate-specific antigen (PSA) in men genetically predisposed to developing prostate cancer. Furthermore, comparing the results of studies that have examined the efficacy of screening for prostate cancer is difficult because studies vary with regard to the cutoff values chosen for an elevated PSA test. For a given sensitivity and specificity of a screening test, the positive predictive value (PPV) increases as the underlying prevalence of disease rises. Therefore, it is theoretically possible that the PPV and diagnostic yield will be higher for the DRE and for PSA in men with a genetic predisposition than in average-risk populations.[1,2]
Most retrospective analyses of prostate cancer screening cohorts have reported PPV for PSA, with or without DRE, among high-risk men in the range of 23% to 75%.[2,3,4,5,6] Screening strategies (frequency of PSA measurements or inclusion of DRE) and PSA cutoff for biopsy varied among these studies, which may have influenced this range of PPV. Cancer detection rates among high-risk men have been reported to be in the range of 4.75% to 22%.[2,5,6] Most cancers detected were of intermediate Gleason score (5–7), with Gleason scores of 8 or higher being detected in some high-risk men. Overall, there is limited information about the net benefits and harms of screening men at higher risk of prostate cancer. In addition, there is little evidence to support specific screening approaches in prostate cancer families at high risk. Risks and benefits of routine screening in the general population are discussed in Prostate Cancer Screening. On the basis of the available data, most professional societies and organizations recommend that high-risk men engage in shared decision-making with their health care providers and develop individualized plans for prostate cancer screening based on their risk factors. A summary of prostate cancer screening recommendations for high-risk men by professional organizations is shown in Table 7 and Table 8.
| | Age to Begin PSA Screening | Screening Interval |
|---|---|---|
| PSA = prostate-specific antigen. | ||
| a Forgermline pathogenic variantsother thanBRCA2(includingATMand Lynch syndromegenes), it is reasonable to consider beginning shared decision-making about PSA screening at age 40 years and to consider screening at annual intervals, rather than every other year.[7] | ||
| BRCA1Carriers | Consider screening[8]or shared-decision making about screening[7]at age 40 years or 10 years before the youngest prostate cancer diagnosis in the family[8] | Consider annual screening rather than screening every other year[7] |
| BRCA2 Carriers | Recommend screening at age 40 years[7,8]or 10 years before the youngest prostate cancer diagnosis in the family[8] | Consider annual screening rather than screening every other year[7] |
| HOXB13 Carriers | Consider shared-decision making about screening at age 40 years[7] | Consider annual screening rather than screening every other year[7] |
| Screening Recommendation Source | Population | Test | Age Screening Initiated | Frequency | Comments |
|---|---|---|---|---|---|
| DRE = digital rectal exam; FDR = first-degree relative; NCCN = National Comprehensive Cancer Network; PSA = prostate-specific antigen; SDR =second-degree relative. | |||||
| a DRE is recommended in addition to PSA test for men with hypogonadism. | |||||
| b A suspicious family history includes, but is not limited to, an FDR or SDR with metastatic prostate cancer, ovarian cancer, male breast cancer, female breast cancer at age ≤45 y, colorectal or endometrial cancer at age ≤50 y, or pancreatic cancer; this may also include two or more FDRs or SDRs with breast, prostate (excluding clinically localized Grade Group 1 disease), colorectal, or endometrial cancer at any age. | |||||
| United States Preventive Services Task Force (2018)[9] | Men aged 55–69 y | PSA | N/A | N/A | In determining whether PSA-based screening is appropriate in individual cases, patients and clinicians should consider the benefits and harms of PSA screening based on family history, race and ethnicity, comorbid medical conditions, patient values about the benefits and harms of screening and treatment-specific outcomes, and other health needs |
| American Urological Association (2023)[10] | African American men, men with germline pathogenic variants in hereditary prostate cancer genes, and men with strong family histories of prostate cancer | PSA | 40 to 45 y | Screening is individualized based on the patient's personal preferences and an informed discussion regarding the uncertainty of benefit and associated harms | |
| American Cancer Society (2023)[11] | African American men | PSA with or without DREa | ≥45 y | Screen every 2 y if PSA is <2.5 ng/mL; screen annually if PSA level is ≥2.5 ng/mL; if PSA levels are between 2.5–4.0 ng/mL, an individualized risk assessment can be performed, which incorporates other prostate cancer risk factors (particularly for high-grade cancer, which may be used for a referral recommendation) | Counseling consists of a review of the benefits and limitations of testing so that a clinician-assisted, informed decision about testing can be made. It is recommended that prostate cancer screening be accompanied by an informed decision-making process |
| Men with an FDR who was diagnosed with prostate cancer at <65 y | PSA with or without DREa | ≥45 y | |||
| Men with multiple FDRs who were diagnosed with prostate cancer at <65 y | PSA with or without DREa | ≥40 y | |||
| NCCN Prostate Cancer Early Detection (Version 2.2023)[7] | African American men | Baseline PSA | 40 y | Consider screening at annual intervals rather than every other year | The panel states that it is reasonable for African American men to consider beginning shared decision-making about PSA screening with their providers at age 40 y |
| Men with a suspicious family historyb | Baseline PSA | 40 y | Screen every 2–4 y if PSA level <1 ng/mL, DRE normal; if the family history is concerning, NCCN recommends shared decision-making to determine the frequency of PSA screening | Referral to a cancer genetics professional is recommended for those with a known or suspected pathogenic variant in a cancersusceptibility gene [7] | |
| Screen every 1–2 y if PSA level ≤3 ng/mL, DRE normal (if done) | |||||
Level of evidence: 5
Screening in carriers ofBRCApathogenic variants
IMPACT (Identification of Men with a genetic predisposition to ProstAte Cancer) is an international study focused on prostate cancer screening in carriers of BRCA1/BRCA2 pathogenic variants versus noncarriers.[12] The study recruited 2,481 men (791 BRCA1 carriers, 531 BRCA1 noncarriers; 731 BRCA2 carriers, 428 BRCA2 noncarriers). A total of 199 men (8%) presented with PSA levels higher than 3.0 ng/mL, which was the study PSA cutoff for recommending a biopsy. The overall cancer detection rate was 36.4% (59 prostate cancers diagnosed among 162 biopsies). Prostate cancer by BRCA pathogenic variant status was as follows: BRCA1 carriers (n = 18), BRCA1 noncarriers (n = 10); BRCA2 carriers (n = 24), BRCA2 noncarriers (n = 7). Using published stage and grade criteria for risk classification,[13] intermediate- or high-risk tumors were diagnosed in 11 of 18 BRCA1 carriers (61%), 8 of 10 BRCA1 noncarriers (80%), 17 of 24 BRCA2 carriers (71%), and 3 of 7 BRCA2 noncarriers (43%). The PPV of PSA with a biopsy threshold of 3.0 ng/mL was 48% in carriers of BRCA2 pathogenic variants, 33.3% in BRCA2 noncarriers, 37.5% in BRCA1 carriers, and 23.3% in BRCA1 noncarriers. Ninety-five percent of the men were White; therefore, the results cannot be generalized to all ethnic groups.
Interim results from the IMPACT study (now comprising 2,932 participants including 919 BRCA1 carriers and 902 BRCA2 carriers) demonstrated a cancer incidence rate (per 1,000 person-years) that was higher in BRCA2 carriers compared with noncarriers (19 vs. 12; P = .03). There was no statistical difference in the cancer incidence rates between BRCA1 carriers and noncarriers. Cancer in BRCA2 carriers, but not in BRCA1 carriers, was diagnosed at an earlier age and was more likely to be clinically significant.[14]
Level of evidence (screening in carriers of BRCA pathogenic variants): 3
Impact of Germline Genetics on Management and Treatment of Metastatic Prostate Cancer
Targeted therapies on the basis of genetic results are increasingly driving options and strategies for treatment in oncology. These therapeutic approaches include candidacy for targeted therapy (such as poly [ADP-ribose] polymerase [PARP] inhibitors or immune checkpoint inhibitors), use of platinum-based chemotherapy, and sequencing of androgen-signaling therapy versus chemotherapy. Multiple genetically informed clinical trials are under way for men with prostate cancer.[15]Table 9 summarizes some of the published precision oncology and precision management studies.
| Study | Cohort | Germline Results | Intervention | Outcomes and Comments | |
|---|---|---|---|---|---|
| ADT = androgen deprivation therapy; AR = androgen receptor; CI = confidence interval; CSS = cause-specific survival; DDR = DNA damage repair; FDA = U.S. Food and Drug Administration; HR = hazard ratio; HRR = homologous recombination repair; mCRPC = metastatic castration-resistant prostate cancer; mPC = metastatic prostate cancer; ORR = objective response rate; OS = overall survival; PARP = poly (ADP-ribose) polymerase; PC = prostate cancer; PFS = progression-free survival; PSA = prostate-specific antigen; RR = relative risk. | |||||
| a This study reported both germline and somatic genetic test results. | |||||
| Retrospective | |||||
| Annala et al. (2017)[16] | 319 men with mCRPC; performed germline sequencing of 22 DNA repair genes; all participants previously received ADT and their PCs progressed | 24/319 (7.5%) had DDR germline pathogenic variants: | Patientswith mCRPC and a germline pathogenic variant received the following as a first-line AR-targeted therapy: docetaxel/cabazitaxel (41%), enzalutamide (23%), or abiraterone (36%) | Patients with DNA repair defects had decreased responses to ADT: | |
| —BRCA2: 16/319 (5.0%) | |||||
| —ATM: 1/319 (0.3%) | — Time from ADT initiation to mCRPC: Germline positive, 11.8 mo (n = 22) vs. germline negative, 19.0 mo (n = 113) (P = .031) | ||||
| —BRCA1: 1/319 (0.3%) | Patients with mCRPC butwithout a germline pathogenic variant received the following as a first-line AR-targeted therapy: docetaxel/cabazitaxel (33%), enzalutamide (18%), abiraterone (39%), or other (10%) | ||||
| —PALB2: 2/319 (0.6%) | — PFS on first-line AR-targeted therapy: Germline positive, 3.3 mo vs. germline negative, 6.2 mo (P = .01) | ||||
| Pomerantz et al. (2017)[17] | 141 men with mCRPC treated with docetaxel | 8/141 (5.7%) hadBRCA2germline pathogenic variants | Patients received at least two doses of carboplatin and docetaxel | 6/8 men withBRCA2germline pathogenic variants (75%) had PSA levels that declined by 50% vs. 23/133 in men withoutBRCA2germline pathogenic variants (17%) (P< .001) | |
| A small case series (n = 3) showed a response to platinum chemotherapy with biallelic inactivation ofBRCA2, defined as either biallelic somaticBRCA2pathogenic variants or a germline pathogenic variant plus a somaticBRCA2pathogenic variant[18] | |||||
| Mateo et al. (2018)[19] | 390 men with mPC; retrospective review | 60/390 (15.4%) had DDR germline pathogenic variants: | Patients received abiraterone, enzalutamide, and docetaxel; an exploratory subgroup analysis was done for PARP inhibitors/platinum chemotherapy | Similar findings were observed for DDR pathogenic variant carriers and noncarriers for several outcome measures: | |
| — Median OS from castration resistance (3.2 y in carriers vs 3.0 y in noncarriers;P = .73) | |||||
| — Median docetaxel PFS (6.8 mo in carriers vs. 5.1 mo in noncarriers) | |||||
| —BRCA2: 37/390 (9.5%) | — RRs for PC (61% in carriers vs. 54% in noncarriers) | ||||
| — Median PFS on first-line abiraterone/enzalutamide (8.3 mo in both carriers and noncarriers) | |||||
| — RR of PC on first-line abiraterone/enzalutamide (46% in carriers vs. 56% in noncarriers) | |||||
| Carter et al. (2019)[20] | 1,211 men with PC on active surveillance | 2.1% of patients had germline pathogenic variants inBRCA1/BRCA2/ATM | Patients were put on active surveillance | 289 patients had their PC tumor grades reclassified: 11/26 patients had pathogenic variants inBRCA1/BRCA2/ATMand 278/1,185 patients did not have a pathogenic variant inBRCA1/BRCA2/ATM(noncarriers); adjusted HR, 1.96 (95% CI, 1.004–3.84;P = .04) | |
| Tumor reclassification occurred in 6/11BRCA2carriers and 283/1,200 noncarriers; adjusted HR, 2.74 (95% CI, 1.26–5.96;P = .01) | |||||
| Of the men who had their PCs reclassified, 3.8% had aBRCA1,BRCA2, orATMpathogenic variant, and 2.1% only had aBRCA2 pathogenic variant. Of the men whose PCs were not reclassified, 1.6% had aBRCA1,BRCA2, orATMpathogenic variant, and 0.5% only had aBRCA2 pathogenic variant. TheP value forBRCA1/BRCA2/ATMcarriers with PCs reclassified versus those without PCs reclassified was .04. TheP value forBRCA2carriers with PCs reclassified versus those without PCs reclassified was .03 | |||||
| Marshall et al. (2019)[21] | 46 men with mCRPC were offered olaparib; 23 men had germline pathogenic variants (13 men were not tested) | 23 men had germline pathogenic variants inBRCA1/BRCA2/ATM; 2 men hadBRCA1pathogenic variants, 15 men hadBRCA2 pathogenic variants, and 6 men hadATMpathogenic variants | Patients received olaparib | When patients were given olaparib, PSA levels were reduced by 50% in 13/17 (76%) men withBRCA1/BRCA2pathogenic variants and in 0/6 (0%) men withATMpathogenic variants (Fisher's exact test;P = .002) | |
| Patients withBRCA1/BRCA2 pathogenic variants had a median PFS of 12.3 mo, while patients withATMpathogenic variants had a median PFS of 2.4 mo (HR, 0.17; 95% CI, 0.05–0.57;P = .004) | |||||
| Sokolova et al. (2021)[22] | 90 men with PC; 76/90 had metastatic disease when their PC was diagnosed; participants were matched for PC stage and year of germline testing; participants had similar ages, Gleason grades, and PSA levels at diagnosis | 45 men withATMgermline pathogenic variants; 45 men withBRCA2 germline pathogenic variants | Patients received various systemic therapies | No changes were observed when different groups were given abiraterone, enzalutamide, or docetaxel | |
| When patients were given PARP inhibitors, PSA levels were reduced by 50% in 0/7 men withATMgermline pathogenic variants and in 12/14 men withBRCA2germline pathogenic variants (P< .001); this response was significant | |||||
| Study limitations included the following: retrospective study, no zygosity data | |||||
| Prospective | |||||
| Antonarakis et al. (2018)[23] | 172 men with mCRPC began treatment with abiraterone or enzalutamide | 22/172 (12.8%) had DDR germline pathogenic variants: | Patients received first-line hormonal therapy (abiraterone or enzalutamide) | In propensity score–weighted multivariable analyses, outcomes were superior in men with germlineBRCA1/BRCA2/ATMvariants with respect to PSA-PFS (HR, 0.48; 95% CI, 0.25–0.92;P = .027), PFS (HR, 0.52; 95% CI, 0.28–0.98;P = .044), and OS (HR, 0.34; 95% CI, 0.12–0.99;P = .048). These results were not observed for men with non-BRCA1/BRCA2/ATMgermline variants (P> .10) | |
| —BRCA1/BRCA2/ATM: 9/172 (5.2%) | Study limitations included the following: only 9 patients withBRCA1/BRCA2/ATMpathogenic variants | ||||
| Castro et al. (2019)[24] | 419 men with mCRPC were enrolled when they were diagnosed with mPC | 68/419 (16.2%) had DDR germline pathogenic variants: | Patients received an androgen-signaling inhibitor (abiraterone or enzalutamide) as a first-line therapy and a taxane (docetaxel was given in 96.3% of patients) as a second-line therapyor patients received a taxane as a first-line therapy and an androgen-signaling inhibitor (abiraterone or enzalutamide) as a second-line therapy | CSS betweenATM/BRCA1/BRCA2/PALB2carriers and noncarriers was not statistically significant (23.3 mo vs. 33.2 mo;P = .264) | |
| —BRCA2: 14/419 (3.3%) | |||||
| —ATM: 8/419 (1.9%) | CSS was halved inBRCA2carriers (17.4 mo vs. 33.2 mo;P = .027), andBRCA2pathogenic variants were identified as an independent prognostic factor for CSS (HR, 2.11;P = .033) | ||||
| —BRCA1: 4/419 (1%) | Significant interactions betweenBRCA2status and treatment type (androgen-signaling inhibitor vs. taxane therapy) were observed (CSS-adjustedP = .014; PFS-adjustedP = .005) | ||||
| —PALB2: None | CSS (24.0 mo vs. 17.0 mo) and PFS (18.9 mo vs. 8.6 mo) were greater inBRCA2carriers treated with first-line abiraterone or enzalutamide when compared with first-line taxanes | ||||
| de Bono et al. (2020)[25] | 387 men in the PROfound study who had mCRPC with disease progression while receiving a new hormonal agent (e.g., enzalutamide or abiraterone) | Currently, the FDA has approved olaparib for use in patients with mCRPC who have a somatic or germline pathogenic variant in an HRR gene. The PROfound study cited data fromMateo et al. 2015, which discovered that about half of the HRR gene variants in patient tumors were germline in nature. Results in this study reported on olaparib response in individuals with somatic variants. Data on germline pathogenic variants will be reported in the future | Randomized, open-label, phase III trial in which patients received olaparib (300 mg twice per day)or the physician's choice of enzalutamide (160 mg once per day) or abiraterone (1,000 mg once per day) plus prednisone (5 mg twice per day) | In cohort A, imaging-based PFS was significantly longer in the olaparib group than in the control group (median, 7.4 mo vs. 3.6 mo; HR for progression or death, 0.34; 95% CI, 0.25–0.47;P< .001). The median OS in cohort A was 18.5 mo in the olaparib group and 15.1 mo in the control group; 81% of the patients in the control group who had disease progression crossed over to receive olaparib | |
| Cohort A: 245 men with >1 somatic variant inBRCA1,BRCA2, orATM | |||||
| Cohort B: 142 men with >1 somatic variant in any of the following genes:BRIP1,BARD1,CDK12,CHEK1,CHEK2,FANCL,PALB2,PPP2R2A,RAD51B,RAD51C,RAD51D, orRAD54L | |||||
| Hussain et al. (2020)[26] | 387 men with mCRPC in the PROfound study; PC progressed when taking enzalutamide, abiraterone, or both | Currently, the FDA has approved olaparib for use in patients with mCRPC who have a somatic or germline pathogenic variant in an HRR gene. The PROfound study cited data fromMateo et al. 2015, which discovered that about half of the HRR gene variants in patient tumors were germline in nature. Results in this study reported on olaparib response in individuals with somatic variants. Data on germline pathogenic variants will be reported in the future | Patients received treatment that was randomly assigned in a 2:1 ratio for olaparib versus control therapy; control therapy consisted of the provider's choice of enzalutamide or abiraterone, plus prednisone. Crossover to olaparib was permitted when PC progressed on imaging | The median OS in cohort A was 19.1 mo with olaparib and 14.7 mo with control therapy. The HR for death (adjusted for crossover from control therapy) was 0.42 (95% CI, 0.19–0.91) | |
| Cohort A: 245 men with >1 somatic variant inBRCA1,BRCA2, orATM | The median OS in cohort B was 14.1 mo for olaparib and 11.5 mo for control therapy. The HR for death (adjusted for crossover from control therapy) was 0.83 (95% CI, 0.11–5.98) | ||||
| Cohort B: 142 men with >1 somatic variant in any of the following genes:BRIP1,BARD1,CDK12,CHEK1,CHEK2,FANCL,PALB2,PPP2R2A,RAD51B,RAD51C,RAD51D, orRAD54L | |||||
| Abida et al. (2020)a[27] | 115 men with mCRPC from the TRITON2 study with a deleterious somatic or germline pathogenic variant inBRCA1/BRCA2; patients had mCRPCs that progressed after treatment with one to two lines of next-generation AR-directed therapy and one taxane-based chemotherapy | 44/115 (38%) hadBRCA1/BRCA2germline pathogenic variants: | Patients received one or more doses of rucaparib (600 mg) | The ORR was 43.5% in men with measurable disease and 50.8% in men without measurable disease. ORRs were similar for men with germline and somatic variants and for men withBRCA1/BRCA2pathogenic variants | |
| —BRCA1: 5/115 (4%) | |||||
| —BRCA2: 39/115 (34%) | |||||
| 71/115 (62%) hadBRCA1/BRCA2somatic variants: | 63/115 men had a confirmed PSA response (54.8%), which differed by gene; however, theBRCA1group was small: | ||||
| —BRCA1: 8/115 (7%) | — BRCA1: 2/13 (15.4%) | ||||
| —BRCA2: 63/115 (55%) | —BRCA2: 61/102 (59.8%) | ||||
| De Bono et al. (2021)a[28] | 104 men with progressive mCRPC and pathogenic variants in DDR-HRR genes; patients received at least one dose of talazoparib | 25/71 (25%) patients had germline pathogenic variants: 13 inBRCA2, 4 inATM, and 8 in other genes | Patients received one or more doses of talazoparib per day (received 1 mg per day or 0.75 mg per day if the patient had moderate renal impairment) | The ORR was observed in 7/28 (25%) men with germline pathogenic variants | |
| Patients also had somatic variants in the following genes: 61 inBRCA1/2, 57 inBRCA2, 4 inPALB2, 17 inATM, 22 in other genes (ATR,CHEK2,FANCA,MLH1,MRE11A,NBN, andRAD51C) | After a median follow-up period of 16.4 mo (range, 11.1–22.1), the ORR for patients with somatic variants was 29.8% (31 of 104 patients; 95% CI, 21.2%–39.6%). Clinical benefit (defined as patients with complete response, partial response, or stable disease for ≥6 months from treatment start) varied between individuals with different pathogenic variants:BRCA1/2(56%),BRCA2(56%),PALB2(25%),ATM(24%), other (0%) | ||||
References:
- Sartor O: Early detection of prostate cancer in African-American men with an increased familial risk of disease. J La State Med Soc 148 (4): 179-85, 1996.
- Matikainen MP, Schleutker J, Mörsky P, et al.: Detection of subclinical cancers by prostate-specific antigen screening in asymptomatic men from high-risk prostate cancer families. Clin Cancer Res 5 (6): 1275-9, 1999.
- Catalona WJ, Antenor JA, Roehl KA, et al.: Screening for prostate cancer in high risk populations. J Urol 168 (5): 1980-3; discussion 1983-4, 2002.
- Valeri A, Cormier L, Moineau MP, et al.: Targeted screening for prostate cancer in high risk families: early onset is a significant risk factor for disease in first degree relatives. J Urol 168 (2): 483-7, 2002.
- Narod SA, Dupont A, Cusan L, et al.: The impact of family history on early detection of prostate cancer. Nat Med 1 (2): 99-101, 1995.
- Giri VN, Beebe-Dimmer J, Buyyounouski M, et al.: Prostate cancer risk assessment program: a 10-year update of cancer detection. J Urol 178 (5): 1920-4; discussion 1924, 2007.
- National Comprehensive Cancer Network: NCCN Clinical Practice Guidelines in Oncology: Prostate Cancer Early Detection. Version 2.2023. Plymouth Meeting, Pa: National Comprehensive Cancer Network, 2023. Available online with free registration. Last accessed November 30, 2023.
- National Comprehensive Cancer Network: NCCN Clinical Practice Guidelines in Oncology: Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic. Version 2.2024. Plymouth Meeting, Pa: National Comprehensive Cancer Network, 2023. Available online with free registration. Last accessed October 31, 2023.
- U.S. Preventative Services Task Force: Screening for prostate cancer: U.S. Preventive Services Task Force recommendation statement. Rockville, Md: U.S. Preventative Services Task Force, 2018. Available online. Last accessed November 8, 2023.
- Wei JT, Barocas D, Carlsson S, et al.: Early Detection of Prostate Cancer: AUA/SUO Guideline Part I: Prostate Cancer Screening. J Urol 210 (1): 46-53, 2023.
- American Cancer Society: American Cancer Society Recommendations for Prostate Cancer Early Detection. American Cancer Society, 2023. Available online. Last accessed January 12, 2024.
- Bancroft EK, Page EC, Castro E, et al.: Targeted prostate cancer screening in BRCA1 and BRCA2 mutation carriers: results from the initial screening round of the IMPACT study. Eur Urol 66 (3): 489-99, 2014.
- National Collaborating Centre for Cancer (UK): Prostate Cancer: Diagnosis and Treatment. Cardiff, UK: National Collaborating Centre for Cancer, 2008. Available online. Last accessed November 8, 2023.
- Page EC, Bancroft EK, Brook MN, et al.: Interim Results from the IMPACT Study: Evidence for Prostate-specific Antigen Screening in BRCA2 Mutation Carriers. Eur Urol 76 (6): 831-842, 2019.
- Carlo MI, Giri VN, Paller CJ, et al.: Evolving Intersection Between Inherited Cancer Genetics and Therapeutic Clinical Trials in Prostate Cancer: A White Paper From the Germline Genetics Working Group of the Prostate Cancer Clinical Trials Consortium. JCO Precis Oncol 2018: , 2018.
- Annala M, Struss WJ, Warner EW, et al.: Treatment Outcomes and Tumor Loss of Heterozygosity in Germline DNA Repair-deficient Prostate Cancer. Eur Urol 72 (1): 34-42, 2017.
- Pomerantz MM, Spisák S, Jia L, et al.: The association between germline BRCA2 variants and sensitivity to platinum-based chemotherapy among men with metastatic prostate cancer. Cancer 123 (18): 3532-3539, 2017.
- Cheng HH, Pritchard CC, Boyd T, et al.: Biallelic Inactivation of BRCA2 in Platinum-sensitive Metastatic Castration-resistant Prostate Cancer. Eur Urol 69 (6): 992-5, 2016.
- Mateo J, Cheng HH, Beltran H, et al.: Clinical Outcome of Prostate Cancer Patients with Germline DNA Repair Mutations: Retrospective Analysis from an International Study. Eur Urol 73 (5): 687-693, 2018.
- Carter HB, Helfand B, Mamawala M, et al.: Germline Mutations in ATM and BRCA1/2 Are Associated with Grade Reclassification in Men on Active Surveillance for Prostate Cancer. Eur Urol 75 (5): 743-749, 2019.
- Marshall CH, Sokolova AO, McNatty AL, et al.: Differential Response to Olaparib Treatment Among Men with Metastatic Castration-resistant Prostate Cancer Harboring BRCA1 or BRCA2 Versus ATM Mutations. Eur Urol 76 (4): 452-458, 2019.
- Sokolova AO, Marshall CH, Lozano R, et al.: Efficacy of systemic therapies in men with metastatic castration resistant prostate cancer harboring germline ATM versus BRCA2 mutations. Prostate 81 (16): 1382-1389, 2021.
- Antonarakis ES, Lu C, Luber B, et al.: Germline DNA-repair Gene Mutations and Outcomes in Men with Metastatic Castration-resistant Prostate Cancer Receiving First-line Abiraterone and Enzalutamide. Eur Urol 74 (2): 218-225, 2018.
- Castro E, Romero-Laorden N, Del Pozo A, et al.: PROREPAIR-B: A Prospective Cohort Study of the Impact of Germline DNA Repair Mutations on the Outcomes of Patients With Metastatic Castration-Resistant Prostate Cancer. J Clin Oncol 37 (6): 490-503, 2019.
- de Bono J, Mateo J, Fizazi K, et al.: Olaparib for Metastatic Castration-Resistant Prostate Cancer. N Engl J Med 382 (22): 2091-2102, 2020.
- Hussain M, Mateo J, Fizazi K, et al.: Survival with Olaparib in Metastatic Castration-Resistant Prostate Cancer. N Engl J Med 383 (24): 2345-2357, 2020.
- Abida W, Patnaik A, Campbell D, et al.: Rucaparib in Men With Metastatic Castration-Resistant Prostate Cancer Harboring a BRCA1 or BRCA2 Gene Alteration. J Clin Oncol 38 (32): 3763-3772, 2020.
- de Bono JS, Mehra N, Scagliotti GV, et al.: Talazoparib monotherapy in metastatic castration-resistant prostate cancer with DNA repair alterations (TALAPRO-1): an open-label, phase 2 trial. Lancet Oncol 22 (9): 1250-1264, 2021.
Introduction
The psychological impact of a family history of prostate cancer and/or a positive genetic test for hereditary prostate cancer may influence well-being and screening/prevention behaviors. Important psychosocial issues that have been investigated include perceived risk of prostate cancer, distress, and prostate cancer screening behaviors. Most of this evidence is based on hereditary risk from family history, rather than the results of genetic testing. If known, this section includes data from studies of men who tested positive for hereditary prostate cancer genes. The presence of a prostate cancer family history is important, since most cases of hereditary prostate cancer have unknown etiologies, are polygenic, or cannot be explained by clinical multigene panel tests.[1] For more information about polygenic risk, see the Polygenic risk scores for prostate cancer section.
Prostate Cancer Risk Perception
Understanding drivers of prostate cancer risk perception is important because it can influence other psychological characteristics and is widely regarded as a predictor of health behaviors. Studies that have analyzed the influence of a family history of prostate cancer on perceived cancer risk have had mixed results.
Although family histories of prostate cancer can increase perceived prostate cancer risk in some men,[2] other studies found that men with family histories of prostate cancer considered their risk to be the same as, or less than, that of the average man.[3,4] Other factors, including being married, were associated with increased prostate cancer risk perception.[5] Perceived risk may be positively correlated with levels of concern about developing prostate cancer,[3] depression,[6] and/or the number of relatives who were diagnosed with prostate cancer in a family.[2,3] Confusion regarding the differences between benign prostatic hyperplasia and prostate cancer are confounders in prostate cancer risk perception.[6]
An international study of men with personal and/or family histories of BRCA1/BRCA2 pathogenic variants found that risk perception was associated with intrusive thoughts, avoidance coping, prostate cancer–related anxiety, and worry about prostate cancer.[7]
Psychological Distress
Although up to 50% of first-degree relatives (FDRs) of prostate cancer patients expressed concern about developing prostate cancer in some studies,[3] the level of anxiety reported by these individuals was relatively low and was related to lifetime risk, rather than short-term risk.[3,6] This concern was higher in men who were younger than their FDRs when their prostate cancers were diagnosed.[3] Unmarried FDRs may have worried more about developing prostate cancer than married men did.[3] In a Swedish study, only 3% of participants (n = 110) said that worry about prostate cancer affected their daily lives fairly much, and 28% said that it affected their daily lives slightly.[6]
In men who self-referred for free prostate cancer screening, general– and prostate cancer–related distress did not differ significantly between men who were FDRs of prostate cancer patients and men who were not.[2] In a Swedish study, male FDRs who reported higher levels of worry about developing prostate cancer had higher Hospital Anxiety and Depression Scale (HADS) scores than men with lower levels of worry. In FDRs, the average HADS score was in the 75th percentile.[6]
A study measured anxiety and general quality-of-life in 220 men with family histories of prostate cancer who were undergoing prostate cancer screening with prostate-specific antigen (PSA) tests.[8] In this group, 20% of participants experienced a moderate deterioration in their anxiety scores, and 20% experienced a minimal deterioration in health-related quality-of-life (HRQOL) scores. The average period between assessments was 35 days, which encompassed PSA testing and a wait for results that averaged 15.6 days. Only men with normal PSA values (4 ng/mL or less) were assessed. Factors associated with HRQOL deterioration included being 50 to 60 years old, having more than two relatives with prostate cancer, having an anxious personality, being well-educated, and not having children living at home. The authors stressed that analysis of prostate cancer screening impact on FDRs should not rely solely on mean changes in HRQOL scores. Since a subset of men who received normal results experienced screening-associated distress, interventions may be needed to encourage men with increased hereditary risk to comply with repeated screening requests.
Screening for Prostate Cancer
For more information about prostate cancer screening in the general population, see Prostate Cancer Screening, and for more information about screening individuals with hereditary prostate cancer syndromes, see the Prostate Cancer Screening section.
For most cancer types, knowing that an individual has hereditary risk leads to recommendations for approved (if not proven) screening. This complicates prostate cancer screening, because there is a lack of clear recommendations for many high-risk men and men in the general population. This creates uncertainty about the clinical and psychosocial factors related to prostate cancer screening.
Several small studies have examined the behavioral correlates of prostate cancer screening at average and increased prostate cancer risk, based on family history.[4,6,8,9,10,11,12,13,14] In general, results differed regarding whether men with a family histories of prostate cancer were more likely to be screened than those without hereditary prostate cancer risk. It is unclear if the prostate cancer screening implemented in each group was appropriate for its risk status. Most studies had a relatively small numbers of subjects, and the prostate cancer screening criteria were not uniform across studies, making generalizations difficult. Notably, all of these studies predate the era of hereditary cancer testing, and there is a paucity of research about prostate cancer screening behaviors in males who have undergone hereditary prostate cancer genetic testing.
References:
- Ni Raghallaigh H, Eeles R: Genetic predisposition to prostate cancer: an update. Fam Cancer 21 (1): 101-114, 2022.
- Taylor KL, DiPlacido J, Redd WH, et al.: Demographics, family histories, and psychological characteristics of prostate carcinoma screening participants. Cancer 85 (6): 1305-12, 1999.
- Beebe-Dimmer JL, Wood DP, Gruber SB, et al.: Risk perception and concern among brothers of men with prostate carcinoma. Cancer 100 (7): 1537-44, 2004.
- Miller SM, Diefenbach MA, Kruus LK, et al.: Psychological and screening profiles of first-degree relatives of prostate cancer patients. J Behav Med 24 (3): 247-58, 2001.
- Montgomery GH, Erblich J, DiLorenzo T, et al.: Family and friends with disease: their impact on perceived risk. Prev Med 37 (3): 242-9, 2003.
- Bratt O, Damber JE, Emanuelsson M, et al.: Risk perception, screening practice and interest in genetic testing among unaffected men in families with hereditary prostate cancer. Eur J Cancer 36 (2): 235-41, 2000.
- Bancroft EK, Saya S, Page EC, et al.: Psychosocial impact of undergoing prostate cancer screening for men with BRCA1 or BRCA2 mutations. BJU Int 123 (2): 284-292, 2019.
- Cormier L, Reid K, Kwan L, et al.: Screening behavior in brothers and sons of men with prostate cancer. J Urol 169 (5): 1715-9, 2003.
- Vadaparampil ST, Jacobsen PB, Kash K, et al.: Factors predicting prostate specific antigen testing among first-degree relatives of prostate cancer patients. Cancer Epidemiol Biomarkers Prev 13 (5): 753-8, 2004.
- Bock CH, Peyser PA, Gruber SB, et al.: Prostate cancer early detection practices among men with a family history of disease. Urology 62 (3): 470-5, 2003.
- Jacobsen PB, Lamonde LA, Honour M, et al.: Relation of family history of prostate cancer to perceived vulnerability and screening behavior. Psychooncology 13 (2): 80-5, 2004.
- Roumier X, Azzouzi R, Valéri A, et al.: Adherence to an annual PSA screening program over 3 years for brothers and sons of men with prostate cancer. Eur Urol 45 (3): 280-5; author reply 285-6, 2004.
- Weinrich SP: Prostate cancer screening in high-risk men: African American Hereditary Prostate Cancer Study Network. Cancer 106 (4): 796-803, 2006.
- Ross LE, Uhler RJ, Williams KN: Awareness and use of the prostate-specific antigen test among African-American men. J Natl Med Assoc 97 (7): 963-71, 2005.
The PDQ cancer information summaries are reviewed regularly and updated as new information becomes available. This section describes the latest changes made to this summary as of the date above.
Executive Summary
This section was extensively revised.
Risk Factors for Prostate Cancer
Updated statistics with estimated new cancer cases and deaths for 2024 (cited American Cancer Society as reference 1).
Updated statistics with estimated new cancer cases and deaths for different racial and ethnic groups in 2024.
Risk Assessment for Prostate Cancer
Updated statistics with estimated new cancer cases and deaths for different racial and ethnic groups in 2024 (cited American Cancer Society as reference 1).
Indications for Prostate Cancer Germline Genetic Testing
Added text to state that it is recommended that germline genetic testing candidates undergo genetic education and counseling before participating in testing. Genetic counseling provides information about genetic testing and possible testing outcomes. Genetic education and counseling help individuals make informed decisions about whether they should undergo germline genetic testing.
Genetic Testing Approach for Prostate Cancer
Added text to state that prostate cancer is highly heritable. More than half of an individual's prostate cancer risk is inherited from one's parents (cited Mucci et al. as reference 1). Also added text about inherited prostate cancer risk, which is comprised of many common genetic polymorphisms and rare, deleterious pathogenic variants.
The BRCA1 and BRCA2 subsection was extensively revised.
Added text to state that the clinical utility of genetic testing for the HOXB13 G84E variant is evolving (cited National Comprehensive Cancer Network [Prostate Cancer Early Detection] as reference 21).
Added text about a 2022 study that analyzed prostate cancer risk in 163 men with germline TP53 pathogenic variants (cited Maxwell et al. as reference 72).
The NBN subsection was extensively revised.
Added text about genome-wide association studies (GWAS), which have discovered over 250 prostate cancer risk variants; efforts are being made to integrate these findings into clinical practice via tools like polygenic risk scores.
The Polygenic risk scores for prostate cancer subsection was extensively revised.
Prostate Cancer Genetics: Screening, Surveillance, and Treatment
Table 7, Available Recommendations for Prostate Cancer Screening in BRCA1, BRCA2, and HOXB13 Carriers, was extensively revised.
Added Psychosocial Issues in Familial Prostate Cancer as a new section.
This summary is written and maintained by the PDQ Cancer Genetics Editorial Board, which is editorially independent of NCI. The summary reflects an independent review of the literature and does not represent a policy statement of NCI or NIH. More information about summary policies and the role of the PDQ Editorial Boards in maintaining the PDQ summaries can be found on the About This PDQ Summary and PDQ® Cancer Information for Health Professionals pages.
Purpose of This Summary
This PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about the genetics of prostate cancer. It is intended as a resource to inform and assist clinicians in the care of their patients. It does not provide formal guidelines or recommendations for making health care decisions.
Reviewers and Updates
This summary is reviewed regularly and updated as necessary by the PDQ Cancer Genetics Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH).
Board members review recently published articles each month to determine whether an article should:
- be discussed at a meeting,
- be cited with text, or
- replace or update an existing article that is already cited.
Changes to the summaries are made through a consensus process in which Board members evaluate the strength of the evidence in the published articles and determine how the article should be included in the summary.
The lead reviewers for Genetics of Prostate Cancer are:
- Kathleen A. Calzone, PhD, RN, AGN-BC, FAAN (National Cancer Institute)
- Veda N. Giri, MD (Yale University)
- Suzanne C. O'Neill, PhD (Georgetown University)
- Susan K. Peterson, PhD, MPH (University of Texas, M.D. Anderson Cancer Center)
- Mark Pomerantz, MD (Dana-Farber Cancer Institute)
- John M. Quillin, PhD, MPH, MS (Virginia Commonwealth University)
- Charite Ricker, MS, CGC (University of Southern California)
- Catharine Wang, PhD, MSc (Boston University School of Public Health)
Any comments or questions about the summary content should be submitted to Cancer.gov through the NCI website's Email Us. Do not contact the individual Board Members with questions or comments about the summaries. Board members will not respond to individual inquiries.
Levels of Evidence
Some of the reference citations in this summary are accompanied by a level-of-evidence designation. These designations are intended to help readers assess the strength of the evidence supporting the use of specific interventions or approaches. The PDQ Cancer Genetics Editorial Board uses a formal evidence ranking system in developing its level-of-evidence designations.
Permission to Use This Summary
PDQ is a registered trademark. Although the content of PDQ documents can be used freely as text, it cannot be identified as an NCI PDQ cancer information summary unless it is presented in its entirety and is regularly updated. However, an author would be permitted to write a sentence such as "NCI's PDQ cancer information summary about breast cancer prevention states the risks succinctly: [include excerpt from the summary]."
The preferred citation for this PDQ summary is:
PDQ® Cancer Genetics Editorial Board. PDQ Genetics of Prostate Cancer. Bethesda, MD: National Cancer Institute. Updated <MM/DD/YYYY>. Available at: https://www.cancer.gov/types/prostate/hp/prostate-genetics-pdq. Accessed <MM/DD/YYYY>. [PMID: 26389227]
Images in this summary are used with permission of the author(s), artist, and/or publisher for use within the PDQ summaries only. Permission to use images outside the context of PDQ information must be obtained from the owner(s) and cannot be granted by the National Cancer Institute. Information about using the illustrations in this summary, along with many other cancer-related images, is available in Visuals Online, a collection of over 2,000 scientific images.
Disclaimer
The information in these summaries should not be used as a basis for insurance reimbursement determinations. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page.
Contact Us
More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website's Email Us.
Last Revised: 2024-06-27
Topic Contents
- Executive Summary
- Introduction
- Risk Factors for Prostate Cancer
- Risk Assessment for Prostate Cancer
- Indications for Prostate Cancer Germline Genetic Testing
- Genetic Testing Approach for Prostate Cancer
- Germline Genetics for Prostate Cancer
- Prostate Cancer Genetics: Screening, Surveillance, and Treatment
- Psychosocial Issues in Familial Prostate Cancer
- Latest Updates to This Summary (06 / 27 / 2024)
- About This PDQ Summary
This information does not replace the advice of a doctor. Ignite Healthwise, LLC, disclaims any warranty or liability for your use of this information. Your use of this information means that you agree to the Terms of Use. Learn how we develop our content.